匹伐他汀鈣 Pitavastatin calcium

結構式
| 物競編號 | 0A8Y |
|---|---|
| 分子式 | (C25H23FNO4)2.Ca |
| 分子量 | 880.98 |
| 標簽 | Livalo, 抑制劑 |
編號系統
CAS號:147526-32-7
MDL號:暫無
EINECS號:暫無
RTECS號:暫無
BRN號:暫無
PubChem號:暫無
物性數據
性狀:白色固體。
毒理學數據
暫無
生態學數據
暫無
分子結構數據
暫無
計算化學數據
1.疏水參數計算參考值(XlogP):無
2.氫鍵供體數量:4
3.氫鍵受體數量:12
4.可旋轉化學鍵數量:14
5.互變異構體數量:無
6.拓撲分子極性表面積187
7.重原子數量:63
8.表面電荷:0
9.復雜度:626
10.同位素原子數量:0
11.確定原子立構中心數量:4
12.不確定原子立構中心數量:0
13.確定化學鍵立構中心數量:2
14.不確定化學鍵立構中心數量:0
15.共價鍵單元數量:3
性質與穩定性
暫無
貯存方法
暫無
合成方法
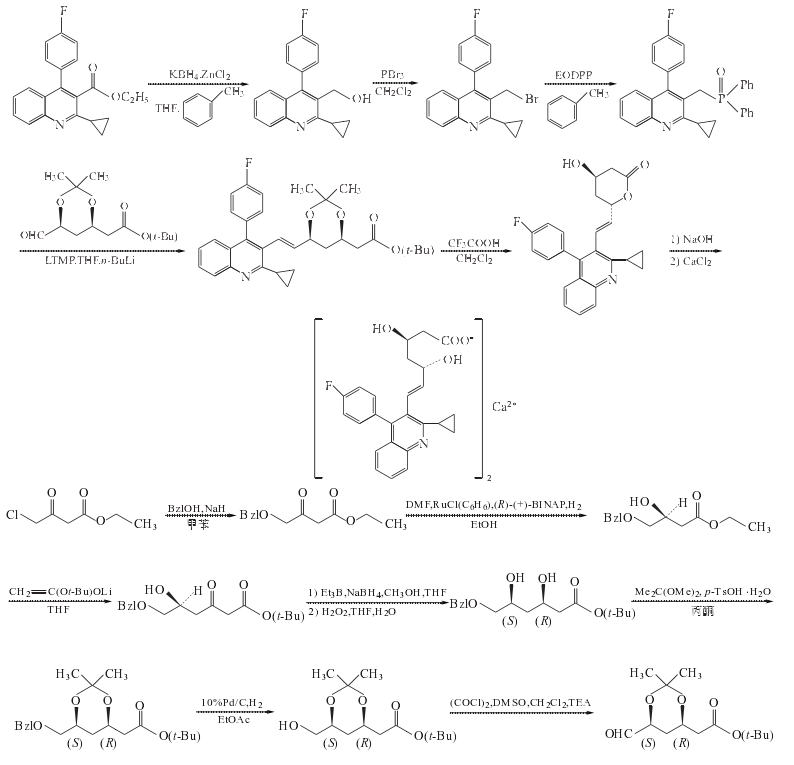
1. 2-環丙基-4-(4-氟苯基)喹啉-3-甲醇的制備
在反應瓶中加入THF196ml、ZnCl2 31.5g(0.23mol)和KBH4 24.5g(0.45mol),室溫攪拌2h.加入2-環丙基-4-(4-氟苯基)喹啉-3-羧酸乙酯49g(0.15mol)的甲苯392ml溶液,緩慢升溫至回流,攪拌回流反應20h.冷至室溫,過濾,濾餅用甲苯加熱回流洗滌,抽濾,合并有機層,加水后靜置分層,水層用甲苯提取,合并有機層,用0.1mol/LNaOH水溶液洗滌,飽和鹽水洗至中性.減壓濃縮至干,剩余物用甲苯重結晶,得2-環丙基-4-(4-氟苯基)喹啉-3-甲醇27.7g,收率65%,mp128~130oC(文獻[4]收率70%,mp136~137oC)
2. 3-溴甲基-2-環丙基-4-(4-氟苯基)喹啉的制備
在反應瓶中加入上步制備的化合物2-環丙基-4-(4-氟苯基)喹啉-3-甲醇25g(85mmol)和二氯甲烷200ml,降溫至0oC,在0oC攪拌滴加三溴化磷(PBr3)16ml(170mmol)的二氯甲烷50ml溶液,溫攪拌反應3h.蒸去二氯甲烷,余物緩慢倒入飽和NaHCO3水溶液中,至pH8~9,乙酸乙酯提取,機相用水洗至中性,壓濃縮至干,余物用無水乙醇重結晶,3-溴甲基-2-環丙基-4-(4-氟苯基)喹啉27g,收率90%mp129~131oC(文獻[6]收率89%,mp140 oC)
3. 2-環丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉的制備
在茄形瓶中加入上步制備的化合物3-溴甲基-2-環丙基-4-(4-氟苯基)喹啉27g(75.8mmol)、乙氧基二苯基膦35ml(150mmol)和甲苯270ml,回流12h.減壓蒸干,剩余物用二氯甲烷/石油醚重結晶,得2-環丙基-3-(二苯基氧膦甲基)-4-(4-氟苯基)喹啉36.0g,收率定量,mp172~174oC.
4. (3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的制備
(1) 芐氧基乙酰乙酸乙酯的制備
在反應瓶中加入甲苯1000ml和55%~60%NaH73.5g(1.68~1.84mol),攪拌懸浮,降溫至20~25oC,往懸浮液滴加苯甲醇162g(155ml,1.498mol),在50min內滴加完,滴加過程中應用冰水浴微微冷卻以保持溫度在20~25oC范圍內,滴畢,將稠厚的漿狀液于24oC繼續攪拌2h,氫氣逸出終止.再在20~25oC攪拌滴加4-氯乙酰乙酸乙酯125g,(103ml,1.7595mol),30min內滴加完,滴加過程中因放熱反應,要適度冷卻,以保持溫度恒定在20~25oC之間.滴畢,繼續在03M攪拌反應19h.反應過程中用GC進行監測(GCl,tR:苯甲醇1.2min,化合物4-氯乙酰乙酸乙酯2.0min,化合物芐氧基乙酰乙酸乙酯7.1min)反應畢,于20~25oC并微冷卻下,在30min內加入2mol/L檸檬酸水溶液755ml(1.51mol),當該溶液加入30%以后,反應液成為非常稠厚漿狀物,繼續加入后,則反應液黏度降低.最后反應混合物pH為4.靜置分層,水溶液層用甲苯100ml提取,合并有機層,用無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑,剩余油狀物加庚烷(25ml×2)一起攪拌,分取庚烷層,仍為油狀的庚烷層真空下濃縮,得粗品225g,將該粗品用雙級薄膜蒸發器進行真空蒸餾[%控制第一蒸餾柱溫140oC,第二蒸餾柱溫180oC,真空度為66.66Pa(0.5Torr),得芐氧基乙酰乙酸乙酯純品150g,收率84%,為無色黏滯油狀物#純度>99%
(2)(S)4-芐氧基-3-羥基丁酸乙酯的制備
在反應瓶中加入新鮮蒸餾的DMF900ml,在氬氣通過液面以下保護下再加入(R)-(+)-2,2’-雙(二苯膦基)-1,1’-聯萘和苯基氯化釕二聚物7.0g(14.0mmol,28.0mmol RU),在氬氣連續通過混合物的條件下,攪拌15min.然后反應瓶深深浸入預先加熱到100oC的油浴中,則溶液澄清,并變成紅棕色清液.將其在氬氣保護下冷至25oC備用.此時,在30L不銹鋼高壓反應釜中加入無水乙醇10L和上步反應制備的化合物芐氧基乙酰乙酸乙酯保持釜內微帶正壓的氬氣保護下加入上述制備的催化劑,加畢,密閉高壓釜,用H2置換釜中氬氣3次.然后充H2,攪拌加熱至內溫100oC此時再通入H2調釜內H2壓力為0.4MPa,恒壓0.4MPa H2壓力和100oC下,一般反應12H則達反應終點.反應畢,降至室溫,通過取樣閥取小樣進行分析,GC3分析表明>99%已氫化.HPLC分析粗品的光學純度為97.4%.用N2將釜內氣體清除3次,反應液放至蒸餾釜,高壓釜用乙醇1L進行清洗,洗液與反應液合并,真空蒸除乙醇,高真空60oC下蒸除DMF.剩余物用薄膜蒸發器于140~150oC蒸餾柱溫/13.33Pa條件下蒸出目的物(S)4-芐氧基-3-羥基丁酸乙酯,得(S)4-芐氧基-3-羥基丁酸乙酯9.70kg,收率96%,為無色油狀物#如果在0~10oC則固化結晶.
(3)(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯的制備
在反應釜中加入二異丙胺9.4L,(71.7mol)和THF13.4L,攪拌下于0~10oC1h內加入15%丁基鋰的己烷溶液43.6L(69.8mol),該混合溶液在10oC攪拌30min,再于-40oC 40min內滴加乙酸叔丁酯9.0ml(66.8mol),再于-40oC 30min內滴加上步制備的化合物(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯4.0kg(16.8mol)的THF4L溶液,將混合物于-40oC攪拌反應1h.用TLC跟蹤確認反應達終點后,往反應混合物中加水(沒有冷卻)7.4L,30min加完,加完水后使反應混合物達-10oC分出有機層,水層用甲苯(16L×2)提取,合并有機層,用無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑[,得半固體粗品(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯7.3~7.7kg,內含有相當量的溶劑.
將粗品(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯的半固體用異丙醚洗滌研磨搗碎后過濾,高真空下干燥,可得純的分析試樣,mp136~137oC.
(4)3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯的制備
在反應釜中加入THF7.5L和上步制備的化合物(S)-6-芐氧基-5-羥基-3-氧代己酸叔丁酯7.54kg(16.8mol),攪拌溶解,20~25oC15min內加入1mol/L三乙基硼的己烷溶液40.0L(40.0mol),反應吸熱.加畢,將溶液攪拌10min.然后冷卻至-60oC,立刻加入硼氫化鈉(NaBH4)1.9kg(50.4mol)(反應不放熱),再在-60oC 2.5h內滴加甲醇35L(強放熱),滴加畢,在同溫度下攪拌1h.TLC跟蹤[展開劑為庚烷/乙酸乙酯(2:1)]確認反應終點,反應畢,15min內加水17L,將反應混合物轉至裝有攪拌器的200L分離器中,加入鹽水17L,分相,水相用甲苯(33L×2)提取,分相后沉淀鹽被沉積在分離器底部.合并有機相,用水22L洗滌#然后加入無水Na2SO4過夜.過濾掉干燥劑Na2SO4后,將濾液真空濃縮.剩余物加THF 34L溶解,在0~10 oC和有效冷卻下加入35%H2O2 7.5L,在1h內加完.將混合物繼續攪拌30min,TLC跟蹤,確認中間體硼酯反應完全,并形成了化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯后,加入水16L和甲苯16L.再將混合物攪拌5min,靜置分相.在有效冷卻下,往分出的有機相加入NaHCO3 2.0kg,破壞過量的H2O2.混合物再攪拌10min.有機相用飽和的NaHCO3水溶液10L洗滌,然后用鹽水5L洗滌,無水Na2SO4干燥,過濾,濾液減壓蒸除溶劑,得油狀剩余物7.0kg,與庚烷12L混合后轉至裝有攪拌器的100L分離器中,將混合物強烈攪拌1h.然后讓其沉降2~3h.從產物相分取庚烷相,庚烷相再加入庚烷24L稀釋,于5 oC攪拌過夜.分取油狀產物相,含在合并產物相的殘留溶劑減壓蒸除,得純的3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯4.2kg(13.5mol),以(S)4-芐氧基-3-羥基丁酸乙酯計收率80%,產物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯為特別淡的黃色油狀物.
(5)(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯的制備
在反應釜中加入上步制備的化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯11.1kg(35.76mol)和丙酮112L,開攪拌,冷至20~25oC,加入2,2-二甲氧基丙烷7.35kg(8.68L,69.2mol)和對甲苯磺酸一水合物0.68kg(3.6mol),將起初的無色溶液于24oC攪拌4h.在攪拌1~2h后溶液慢慢轉變成黃色,用HLC跟蹤,確認化合物3(R),5(S)-6-芐氧基-3,5-二羥基己酸叔丁酯已反應生成產物(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯后,加入固體NaHCO3 2.4kg(28.6mol),將懸浮液攪拌30min.然后將混合物過濾,濾液真空蒸除溶劑,得無色油狀物11.5kg,粗品收率92%.
往200cm×30cm(外徑)(體積141L)MPLC柱裝入硅膠(70~210um)110kg和環己烷/乙酸乙酯(8:1)200L配成的漿料(溢流裝柱).將上述粗品11.5kg溶于環己烷5L中,經過濾除去甲苯磺酸鈉后的濾液進入上述色譜柱分離純化(詳細操作見文獻[9]),經標準后處理和高真空干燥后,得純的化合物(3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯6.9kg,收率69%.純度>99%(HPLC),產物為無色油狀物.
(6) (3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯的制備
在40L的搪瓷高壓釜中加入上步制備的化合物3.25kg(9.27mol)和乙酸乙酯30L,用N2置換溶液中和釜中空氣,再加入Pd/C(10%)催化劑0.32kg,用H2置換釜中N2后充H2(壓力1MPa),攪拌下和恒氫氣壓力(1MPa)下于31~33氫化反應5h,經取樣閥取試樣進行TLC分析[展開劑:庚烷/乙酸乙酯(1:1)](3R,5S)-6-芐氧基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯斑點消失,產物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯斑點明顯,則反應達終點.用N2清除釜內氫氣,過濾,濾液真空濃縮蒸除溶劑,剩余物加甲苯(1L×2),每次都真空蒸除甲苯,剩余物在高真空下干燥,得黏滯無色油狀物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯2.42kg,收率100%,純度>98%.
(7) (3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的制備
在干燥的四口反應瓶中加入干燥的二氯甲烷25ml和草酰氯1.0ml(11mmol),降溫至-50~-60oC,攪拌下慢慢往草酰氯的二氯甲烷溶液滴加DMSO1.7ml(1.72g,22mmol)與二氯甲烷5ml混合液,反應混合物攪拌2min,然后在同溫度下5min內滴加上步制備的化合物(3R,5S)-6-羥基-3,5-O-異亞丙基-3,5-二羥基己酸叔丁酯2.6g(10mmol)與二氯甲烷10ml的混合液,加畢,連續攪拌15min.再加入三乙胺,將混合物攪拌5min.然后升至室溫#加水50ml,攪拌片刻后靜置分層,水層用二氯甲烷50ml反提取.合并有機層,用飽和Na2CO3水溶液洗滌,無水MgSO4干燥,過濾,濾液用旋轉蒸發器濃縮至25ml,依次用1%稀鹽酸、水、5%稀Na2CO3水溶液和水洗滌,然后將有機相濃縮至無溶劑,則得(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯的粗品2.50g,收率97%,可直接用于下步反應.
5. (3R,5S,6E)-7-[2-環丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羥基-3,5-O-異亞丙基-6-庚烯酸叔丁酯的制備
在反應瓶中加入2,2,6,6-四甲基哌啶鋰20ml(120mmol)和THF200ml,0oC和N2保護下,攪拌滴加2.5mol/L正丁基鋰,滴畢,于0oC攪拌15min.冷卻至-78oC,滴加化合物(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯50g(105mmol)的THF800ml溶液,攪拌1h.加入上步制備的中間體(3R,5S)-6-氧代-3,5-二羥基-3,5-O-異亞丙基己酸叔丁酯33.5g(130mmol)的THF200ml溶液,攪拌1h后升至室溫反應過夜.加入飽和NaHCO3水溶液500ml終止反應,用乙酸乙酯提取,提取相用飽和鹽水洗至中性,無水Na2SO4干燥,過濾,濾液減壓蒸干,剩余物經硅膠柱色譜分離,得油狀物37.5g,收率69%.
6. (4R,6S,E)-6-[2-環丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羥基-3,4,5,6-四氫-2H-吡喃-2-酮的制備
在反應瓶中加入上步制備的化合物(3R,5S,6E)-7-[2-環丙基-4-(4-氟苯基)喹啉-3-基]-3,5-二羥基-3,5-O-異亞丙基-6-庚烯酸叔丁酯2.52g(4.9mol)的二氯甲烷20ml溶液,攪拌降溫至0oC,滴加三氟乙酸5.6ml(73.5mmol)的二氯甲烷8ml溶液,滴畢,室溫攪拌反應2h.加入飽和NaHCO3水溶液終止反應.調至pH7~8,用二氯甲烷提取,提取相用水洗滌,無水Na2SO4干燥,過濾,濾液減壓濃縮至干,得1.97g,收率99.5%,mp128~130.
7. (3R,5S,6E)-(+)-7-[2-環丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羥基-6-庚烯酸鈣鹽(2:1)(匹伐他汀鈣)的合成
在反應瓶中加入上步制備的化合物(4R,6S,E)-6-[2-環丙基-4-(4-氟苯基)喹啉-3-基-乙烯基]羥基-3,4,5,6-四氫-2H-吡喃-2-酮1.2g(3.0mmol)和無水乙醇20ml,攪拌溶解,再加入0.1mol/LNaOH水溶液32ml,室溫攪拌反應1h.加入0.1mol/L鹽酸調至pH7,減壓蒸除溶劑,剩余物中加水20ml和無水氯化鈣0.32g(2.9mmol),攪拌15min,后抽濾,濾餅用水洗滌,抽干,烘干,用異丙醇/水重結晶,得白色固體(3R,5S,6E)-(+)-7-[2-環丙基-4-(4-氟苯基)喹啉-3-基] -3,5-二羥基-6-庚烯酸鈣鹽(2:1)(匹伐他汀鈣)1.0g,收率76.3%.
用途
本品為3-羥基-3-甲基戊二酰輔酶A還原酶抑制劑,可以減少肝臟合成膽固醇,使血漿低密度脂蛋白膽固醇的清除增加,可阻止動脈粥樣硬化的進展.口服本品生物利用度高,半衰期長.具有肝臟選擇性,與通過細胞色素P450系統代謝的藥物發生相互作用的傾向更低.其降低甘油三酯的作用強于西立伐他汀、普伐他汀、氟伐他汀和辛伐他汀.臨床上本品用于治療高脂血癥.
安全信息
危險運輸編碼:暫無
危險品標志:暫無
安全標識:暫無
危險標識:暫無
文獻
【1】 四川美康醫藥軟件研究開發有限公司編著.藥物臨床信息參考.成都:四川出版集團四川科學技術出版社,2006:532. 【2】 Aoki T,et al.Arzneim-Forsch Drug Res,1997,47:904-909. 【3】 Miyachi N,et al.Tetrahedrom Lett,1993,34:8267-8270. 【4】 EP,304063.1989. 【5】 Wess G,et al.Tetrahedrom Lett,1990,31:2545-2548. 【6】 日本公開特許93-10700.(CA,1994,120:244704.) 【7】 EP,409281.1991. 【8】 蔡正艷等.中國醫藥工業雜志,2007,38:177-180. 【9】 Beck G,et al.Synthesis,1995,(8):1014-1018. 【10】 Manusco A J,et al.J Org Chem,1978,43:2480-2482. 【11】 Muller P,et al.Terachedrom Lett,1981,22:2361. 【12】 周偉澄主編.高等藥物化學選論.北京:化學工業出版社,2006:314-316. 【13】 Hiyama T,et al.Bull Chem Soc Jpn,1995,68:364-444.
備注
暫無
表征圖譜
暫無圖譜
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號