依非韋侖 Efavirenz
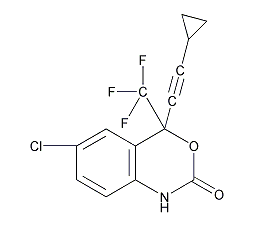
結構式
| 物競編號 | 0059 |
|---|---|
| 分子式 | C14H9ClF3NO2 |
| 分子量 | 315.67 |
| 標簽 | 艾法韋瑞, 施多寧, 依法維侖, (4S)-6-Chloro-4-(2-cyclopropylethynyl)-1,4-dihydro-4-(trifluoromethyl)-2H-3,1-benzoxazin-2-one, 抗真菌感染藥物 |
編號系統
CAS號:154598-52-4
MDL號:暫無
EINECS號:暫無
RTECS號:暫無
BRN號:暫無
PubChem號:暫無
物性數據
1.性狀:白色或類白色結晶粉末
毒理學數據
暫無
生態學數據
暫無
分子結構數據
1、 摩爾折射率:68.40
2、 摩爾體積(cm3/mol):205.2
3、 等張比容(90.2K):549.5
4、 表面張力(dyne/cm):51.3
5、 極化率(10-24cm3):27.11
計算化學數據
1.疏水參數計算參考值(XlogP):4
2.氫鍵供體數量:1
3.氫鍵受體數量:5
4.可旋轉化學鍵數量:1
5.互變異構體數量:2
6.拓撲分子極性表面積38.3
7.重原子數量:21
8.表面電荷:0
9.復雜度:519
10.同位素原子數量:0
11.確定原子立構中心數量:1
12.不確定原子立構中心數量:0
13.確定化學鍵立構中心數量:0
14.不確定化學鍵立構中心數量:0
15.共價鍵單元數量:1
性質與穩定性
暫無
貯存方法
暫無
合成方法
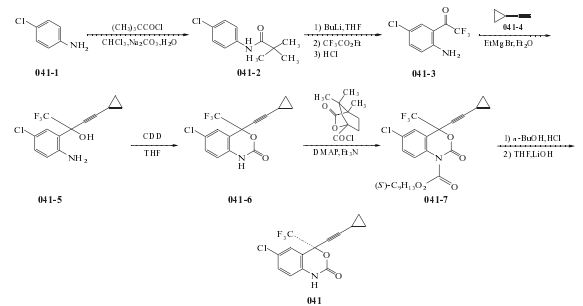
1.對氯苯胺溶于氯仿和飽和碳酸鈉水溶液,滴加2,2-二甲基丙酰氯。加畢,在室溫攪拌,過濾收集固體,濾液分層,分出的氯仿層用鹽水洗,干燥,濃縮。濃縮得到的物質和上述得到的固體一起用煮沸的乙酸乙酯己烷重結晶,得N-2,2-二甲基丙酰基對氯苯胺。將該酰化物溶于四氫呋喃,滴加正丁基鋰的己烷溶液,攪拌,再滴加三氟乙酸乙酯,加入鹽酸和乙酸乙酯,分出有機層,用鹽水洗滌,干燥,減壓濃縮,得到的物質在鹽酸中回流,冷卻,加入乙酸乙酯,用濃氨水調至堿性。分出有機層,用鹽水洗滌,干燥,濃縮,得到2-三氟乙酰基-4-氯苯胺。將環丙基乙炔溶于四氫呋喃,滴加乙基溴化鎂的乙醚溶液進行反應,完畢后,加入2-三氟乙酰基-4-氯苯胺,再滴加飽和的氯化銨水溶液。用乙酸乙酯萃取,萃取液合并后用鹽水洗,干燥,濃縮,得烷化產物。將其和羰基二咪唑溶于無水四氫呋喃。然后減壓蒸出溶劑,加入乙酸乙酯和水。水層用乙酸乙酯萃取。萃取液和有機層合并,用鹽酸、飽和碳酸氫鈉和鹽水洗滌,經干燥、減壓濃縮,得到消旋的依非韋侖。將其和(-)樟腦酰氯在二氯甲烷反應,然后在正丁醇中,加入鹽酸反應,拆分得光學活性的依非韋侖。
2.
合成文獻較多,見文獻[6-12].推薦文獻[6,7]的路線和方法
(1)N-(4-氯苯基)-2,2-二甲基丙酰胺(041-2)的制備
在反應瓶中加入對氯苯胺(041-4)127.57g(1mol)、氯仿1200ml和飽和的Na2CO3水溶液1200ml,攪拌混合,于1h內滴加2,2-二甲基丙酰氯129ml(1.05mol),滴加過程中應劇烈攪拌,滴畢,反應混合物于環境溫度下繼續攪拌反應23h.析出白色晶體過濾收集,濾液轉至分液漏,分取有機層(氯仿層),依次用水和鹽水洗滌,無水MgSO4干燥,過濾,濾液減壓蒸除溶劑,剩余物與上述過濾出的固體合并,用煮沸的乙酸乙酯/己烷重結晶,得化合物041-2 185.6g,收率87.8%,
該化合物為白色結晶性固體.
(2).1-(2-氨基-5-氯苯基)-2,2,2-三氟乙酮(041-3)的制備
往預先干燥好的反應瓶中加入上步制備的化合物041-2 100g(472mmol)和干燥的THF 1000ml,于攪拌下用冰浴冷卻至0 oC,于該溫度下滴加正丁基鋰的己烷溶液(2.5mol/L)387ml(968mmol),滴加過程中保持內溫低于5 oC,1h內滴加畢,在0 oC拌反應1h.在保持反應過程中,反應液中產生橙色沉淀.向該混合物中再滴加1,1,1-三氟乙酸乙酯115ml(968mmol),于1h加完,加畢,繼續攪拌30min.加入5%鹽酸適量終止反應.反應混合物中加入乙酸乙酯1000ml釋,攪拌充分后靜置分層.分取有機層用鹽水洗滌,無水MgSO4干燥,過濾,濾液減壓濃縮,得160g黃色油狀物.將其懸浮于3mol/L鹽酸1000ml中.溶液攪拌回流24h.冷卻,加入乙酸乙酯1000ml稀釋,混合物用濃氨水調至PH呈堿性.靜置分層,取有機層用鹽水洗滌,無水MgSO4干燥,過濾,濾液減壓濃縮,濃縮液經硅膠柱(1.5kg硅膠)色譜分離(洗脫劑:15%乙酸乙酯的己烷溶液),含目的物的洗脫液減壓濃縮至干,得到的固體用煮沸的己烷重結晶,得化合物041-3 57g,率54%,為亮黃色晶體,mp91~92 oC(純品)。
(3)。2-(2-氨基-5-氯苯基)-4-環丙基-1,1,1-三氟-3-丁炔-2-醇(041-5)的制備
在反應瓶中加入THF 250ml和環丙基乙炔(041-4)23g(0.348mol),攪拌溶解,于1h內攪拌下
滴加3mol/L乙基溴化鎂的乙醚溶液116ml(0.348mol).加畢,于0 oC 拌反應1h.然后在40 oC 攪拌反應3h.將反應液再冷卻至0 oC ,在5min內往該反應液中分批加入上步制備的化合物041-3 15.56g(0.0696mol),加畢,在0 oC 拌反應1.5h.再在0 oC 滴加飽和的氯化銨水溶液700ml終止反應.反應混合物用乙酸乙酯(400ml*2)提取,合并有機相,用鹽水洗滌,無水MgSO4干燥,過
濾,濾液經濃縮得到黃色固體,用煮沸的己烷重結晶(沸己烷最終體積100ml),得14.67g,母液濃縮處理可回收041-5 2.1g,合計得041-5 16.77g,mp153~154 oC .
(4).(+-)-6-氯-4-環丙基乙炔基-4-三氟甲基-1,4-二氫-2H-3,1-苯并嗪-2-酮(041-6)的制備
在反應瓶中加入上步制備的化合物041-5 15.00g(0.0518mol)和1,1’-羰基二咪唑(CDD)41.98g(0.259mol)的干燥的THF250ml溶液,在氬氣保護下于55 oC 攪拌反應0C;$利用旋轉蒸發器將反應液中溶劑減壓蒸除,剩余物加入乙酸乙酯500ml和水400ml,分攪拌后靜置分層.水層用乙酸乙酯再提取一次,合并提取液和有機層,用2%鹽酸(200ml*2)洗滌,飽和碳酸氫鈉水溶液洗滌,最后用鹽水洗滌.無水硫酸鎂干燥,過濾,濾液減壓濃縮回收溶劑,得到剩余物16.42g,固體,將其用乙酸乙酯/己烷重結晶得041-6 12.97g品,為白色結晶,mp178~180 oC .
(5)6-氯-1-(1S)-樟腦酰基-4-環丙基乙炔基-4-三氟甲基-1,4-二氫-2H-3,1-苯并嗪-2-酮(041-7)的
制備
在反應瓶中加入上步制備的化合物041-6 (即消旋的Efavirenz)12.97g(0.041mol)、4-二甲基氨基吡啶(DMAP)1.02g(0.0083mol)和(-)樟腦酰氯[(-)-camphanic acid chloride]14.22g(0.06556mol)的干燥二氯甲烷350ml溶液,攪拌,在冰浴冷卻下和氬氣保護下加入三乙胺22.84ml(0.164mol).移去冰浴,自然升至室溫,攪拌反應75min(TLC跟蹤,用4%的乙酸乙酯的氯仿溶液作展開劑,點樣展開,原料斑點消失,反應則基本完成).將反應液用氯仿500ml稀釋,然后用10%的檸檬酸洗滌2次,用水洗1次,鹽水洗1次.無水MgSO4干燥,過濾,濾液減壓濃縮回收溶劑,剩余物為無色泡沫狀物,加入己烷200ml煮沸并研碎,冷卻至室溫,過濾,濾餅用少量冷己烷洗滌,抽干,真空干燥,得041-7 7.79g,白色晶體,mp164~165 oC純度99.2%(HPLC法)
(6)。(4S)-6-氯-4-(環丙基乙炔基)-1,4-二氫-4-(三氟甲基)-2H-3,1-苯并嗪-2-酮(艾法韋瑞)(041)的合成
在反應瓶中加入上步制備的化合物041-7 7.5g(0.01512mol)和正丁醇150ml,在氬氣保護下升溫至60oC攪拌溶解,再加入1mol/L鹽酸10ml,仍保持反應混合物在60oC,攪拌反應72h.用NaHCO3水溶液中和,真空蒸除正丁醇.剩余物用THF150ml溶解,加入2mol/L氫氧化鋰(LiOH)50ml,于室溫攪拌3h.用乙酸乙酯稀釋,攪拌充分后用水洗2次,用鹽水洗滌1次,無水MgSO4干燥,過濾,濾液減壓蒸除溶劑,得白色固體,將其用熱己烷重結晶,得041 3.43g,為白色晶體,mp131~132oC
用途
非核苷類逆轉錄酶抑制劑。與其他病毒逆轉錄酶抑制劑聯合使用,用于HIV-I感染病人的治療。
安全信息
危險運輸編碼:暫無
危險品標志:暫無
安全標識:暫無
危險標識:暫無
文獻
[1] Merck Index. 13th :3552 [2] Young S D,et al,Antimicrob Ag Chemother 1995,39:2602. [3] Staszewski S,et al. N Engl J Med,1999,341:1865. [4]四川美康醫藥軟件研究開發有限公司編著。藥物臨床信息參考。成都:四川出版集團四川科學技術出版社,2006:289-290. [5] 周偉澄主編,高等藥物化學選輪。北京:化學工業出版社,2006:183,187-188 [6]EP,582455.1994. [7]US,5519021.1996. [8]Thompson A S,et al. Tetrahedron Letters,1995,36:8937. [9] Radesca L A,et al.Syn Commum,1997,27:4373. [10]US,5922864.1999. [11]US,6114569.2000. [12]WO,9964405.1999
備注
暫無
表征圖譜

暫無圖譜
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號