化學制備一維無機納米材料的幾種方法
- 2012-12-06
- 專題
自從Iijima[1]發(fā)現(xiàn)了碳納米管,研究學者便熱衷于其他一維納米結(jié)構(gòu)的合成與表征。一維納米結(jié)構(gòu)[2~8]包括納米線、納米棒、納米管、納米帶,由于其特殊的光、電、磁、電化學等性質(zhì),被廣泛應用于催化、電極、電子器件等。在最近幾年里,一系列重要的無機材料納米線被合成及表征,其直徑小至幾納米,長度可達幾微米。而制備納米線最關鍵的因素是控制納米線的尺寸、形狀、結(jié)構(gòu)及其性能。人們采用各種方法[9]合成元素、氧化物、氮化物、碳化物、硫族化合物納米線,合成納米線方法可分為物理方法和化學方法。物理方法采用光、電技術使材料在真空或惰性氣氛中蒸發(fā),然后使原子或分子結(jié)合形成納米線,常用的方法有熱蒸發(fā)、激光燒蝕、機械球磨法等。化學方法一般采用“自下而上”的方法、即通過適當?shù)幕瘜W反應,從分子、原子出發(fā)制備納米材料。在制備無機納米線方面,化學方法顯得更為靈活,有效,可分為溶劑熱、水熱、碳熱反應、化學氣相沉積(CVD) 、前驅(qū)體熱分解法等。所有這些方法都是基于氣相生長、溶液生長兩大類,根據(jù)反應和生長條件的不同,又可細分為8 種。本文介紹了近幾年來化學制備一維無機納米材料的方法,針對各方法列舉了若干實例,并對其未來發(fā)展前景作了展望。
1 無機納米材料合成方法
在一維納米材料的生長中最重要的是納米顆粒的結(jié)晶化過程:從一氣相、液相或固相向另一固相轉(zhuǎn)化,包含著成核和生長兩個過程。當固相的結(jié)構(gòu)單元(原子,離子或分子)的濃度足夠高時,通過均相的成核作用,結(jié)構(gòu)單元集結(jié)成小核或團簇,這些團簇作為晶種使之進一步生長形成更大的團簇。通過控制一維納米線的生長條件,已發(fā)展了多種合成方法,歸結(jié)為氣相生長、溶液生長兩大類。
1.1 氣相生長
氣相生長法可在適宜的氣氛中通過簡單蒸發(fā)技術制備無機材料特別是元素或氧化物納米線,具體可分為以下4 類。
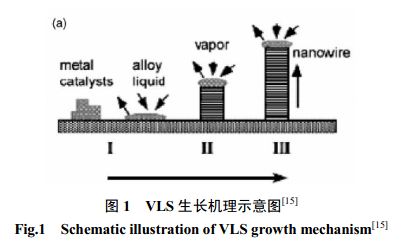
1.1.1 氣-液-固生長(Vapor-liquid-solid growth) 通過含有氣-液-固過程(VLS)的氣相反應生長納米線已受到廣泛研究。單組分納米線的生長一般認為遵循VLS機理[10],即在蒸氣和生長的納米線晶體之間存在由晶體成分與液相生長劑( 或稱催化劑)形成的液相組分,氣相成分首先溶解于液相,并經(jīng)液相進入固相使晶體生長。Wu等[11]在制備Ge納米線的實驗中通過原位透射電鏡觀察證實了VLS生長過程可分為如圖1所示三個階段:金屬合金化、晶體成核和軸向生長。根據(jù)這個機理,在液態(tài)合金或固態(tài)界面存在下可促進各向異性晶體生長。這種機制被廣泛接受并應用于解釋包括Si和Ge在內(nèi)的各種納米線的生長。此種方法合成的納米線的直徑為催化劑顆粒直徑所決定,是獲得均勻尺寸納米線的有效途徑。如在275℃下以Au納米顆粒作為晶種在SiO2/Si基底上通過化學氣相沉積(CVD)可獲得高產(chǎn)量的單晶Ge納米線[12]。據(jù)圖2(a)可知納米線直徑約為25nm,長度可達數(shù)十微米,插圖為CVD之前基底上Au納米顆粒的 AFM照片。由在納米線的末端存在催化劑顆粒的事實表明,該納米線是通過VLS生長機制生長的。通過VLS生長機理于900℃下可制備SiC納米線[13],制得的SiC 納米線的SEM如圖2(b)所示。若通過活性炭和由溶膠-凝膠法制備的表面嵌入Fe 納米顆粒的SiO2反應可制得大量β-SiC納米線[14]。納米線直徑為10~30nm, 長度可達數(shù)十微米。

1.1.2 氧化物協(xié)助生長(Oxide-assisted growth) Lee等[15~16]發(fā)現(xiàn)當Si粉中含有SiO2時,可大大促進Si納米線的生長,于是提出了SiO2促進了Si納米線生長的機理,即氧化物協(xié)助生長機制,與VLS法相比,用此法合成納米線不用金屬催化劑。納米顆粒的成核過程是在底物上發(fā)生的,其反應如下:

在此過程中,熱蒸發(fā)產(chǎn)生的SixO(x>1)蒸汽起了重要作用。這樣的分解導致了Si納米顆粒的沉積,而Si顆粒則成為被SiO2所覆蓋的Si 納米線的核。沉積,成核作用和納米線的生長均發(fā)生在冷的指狀區(qū)域[15,16],說明溫度梯度為納米線的形成和生長提供了外在的作用力。
1.1.3 氣-固生長(Vapor-solid growth) 在采用氣-固生長法(VS 法)制備一維納米晶須過程中,通過蒸發(fā)、化學還原或氣相反應生成蒸汽,蒸汽接下來傳輸并冷凝在底物上。VS法已用于制備 ZnO、SnO2、In2O3、CdO、MgO、Ga2O和Al2O3等氧化物晶須,如果可以控制成核和生長過程,那么采用VS過程合成一維納米材料將有更大的發(fā)展前景。在氧化鋯舟中,通入Ar氣,于1300℃下碳熱反應6h可制得具有納米線和網(wǎng)狀結(jié)構(gòu)的Al2O3納米材料[17]。圖2(c)由以上述過程制得的Al2O3納米結(jié)構(gòu)的SEM圖,此納米結(jié)構(gòu)與納米線相比具有更高的長徑比。此種納米線生長機制可以解釋為VS機制,相關的反應如下:

第一步反應形成低氧化物蒸汽,接著在O2存在下被氧化為Al2O3,而O2可能是系統(tǒng)中本身就存在或是高溫下從氧化鋯中釋放而作為反應物參加反應。
1.1.4 碳熱反應(Carbothermal reaction) 通過碳熱反應可制得一系列氧化物、氮化物、碳化物納米線。以活性炭或碳納米管與氧化物反應先生成低氧化物蒸汽物種,該物種再與C、O2、N2或NH3反應生成所預想的納米線,如在N2或NH3氣中加熱Ga2O3和C的混合物可制備 GaN 納米線[18]。一般情況下碳熱反應涉及以下反應步驟:

以活性炭和硼酸混合物為原料在NH3氣中加熱到1300℃可制備單晶 BN 納米線[19,20]。若在Fe催化劑存在下進行以上實驗,可獲得竹狀納米結(jié)構(gòu)(如圖 3a),竹狀納米 BN外表面上有類似毛發(fā)的形貌特征。這一反應涉及兩個過程:一是碳還原硼酸,二是Fe催化劑促進 BN納米結(jié)構(gòu)的形成。若Fe催化劑分散于SiO2上加熱硼酸則主要得到片狀和須狀的 BN,直徑為60nm,長度可達1μm。可采用碳熱反應制備AlN納米線[21]。將摩爾比為1/1的Al和活性炭放入Al2O3舟或石英舟中置于Al2O3管中在NH3氣氣氛下1300℃加熱5h可制得AlN的納米線,其SEM(如圖 3b)。納米線的長度可達數(shù)微米,直徑在50~100nm 范圍內(nèi)。AlN納米線生長所涉及的反應如下:
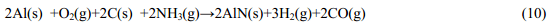
碳在這個反應中對于AlN的形成起了重要作用,碳存在下的 AlN 納米結(jié)構(gòu)的生長是基于VS機制。

1.2 溶液生長法
該納米線生長法可利用各向異性生長,而各向異性生長可通過固體材料結(jié)晶學結(jié)構(gòu)或模板設計等控制。
1.2.1 高度各向異性的晶體結(jié)構(gòu) 固體材料聚硫氮(SN)x 生長為一維納米結(jié)構(gòu),是由結(jié)構(gòu)上各向異性成鍵[22]所決定的。Se[22],Te[23],Mo的硫族化合物[24,25]易獲得納米線,這說明沿著 C-軸結(jié)晶化,是由較強的共價鍵大于鏈間相對較弱的范德華力這一因素所決定的。基于溶液生長機制在適宜的溫度下(110℃)通過加熱回流亞硒酸和肼的混合液,可制得Se納米顆粒,而Se顆粒作為晶種促進Se納米線的生長。圖3c為所制得的三方晶系Se[26]納米線的SEM圖,其直徑在10~800nm范圍內(nèi)可調(diào),長度可達數(shù)百微米。
1.2.2 模板合成 模板合成是制備一維納米結(jié)構(gòu)有效的方法。模板具有納米孔道,如中孔材料、多孔氧化鋁膜、聚碳酸酯膜等。而陽極氧化鋁(AAO)模板合成技術是近年發(fā)展起來的納米結(jié)構(gòu)組裝的最重要技術之一。陽極氧化鋁膜(AAMs)是在酸性介質(zhì)中由陽極氧化鋁片制得的,氧化鋁膜內(nèi)具有均勻尺寸二維的六方圓筒形孔隙(如圖4)[27]。它制作簡單,孔徑大小在5~200nm范圍內(nèi)可調(diào),孔深達幾十到上百微米;本身耐腐蝕、高溫、絕緣;適用于制備金屬(Cu、Au、Bi)、合金(FexAg1-x)、半導體化合物(TiO2、In2O3、CdS、CdSe、Bi2Te3)、導電聚合物、碳納米管等納米組裝材料;去除AAO模板后可得到直徑大小一致的納米粒子、線、棒和管等單分散納米陣列。由于AAO在合成中僅起到一種模板作用,所以在進行納米組裝時,材料的制備仍然需要采用化學反應等途徑來完成。而目前AAO模板的組裝方法主要有電化學沉積法、化學沉積法、化學聚合法、溶膠-凝膠法、化學氣相沉積法等。

通過電沉積和氧化作用在六方形的有序 AAMs 納米孔道上自組裝制備有序In2O3納米線[28]。將8.5g/L InCl3和25g/LNa3C6H5O7·2H2O混合液于室溫下通三探頭直流電將銦納米線電沉積進納米孔洞中。電沉積后,自組裝體系在不同的溫度下于空氣中加熱以形成有序In2O3 納米線陣列。圖5a為在NaOH溶液中蝕去Al后,超聲3min于AAMs模板上生長起來的In2O3納米線陣列的SEM圖。如圖所示,In2O3納米線幾乎具有同樣的尺寸,均勻的嵌在 AAMs 膜上。去除 AAMs 后納米線表面不光滑,直徑分布不均勻。電子衍射分析可知得到的納米線為多晶。采用多孔氧化鋁模板合成排列整齊的單分散的B4C3納米纖維[29],圖5b 顯示納米纖維直徑250nm,長達45μm。XRD和TEM分析表明1000℃下納米纖維變?yōu)闊o定形,1025℃又變?yōu)榫w。

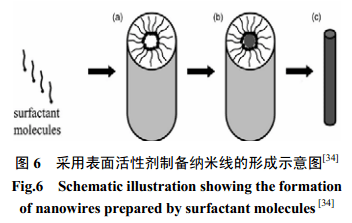
通過表面活性劑自組裝中間相結(jié)構(gòu)也能提供另一組更為有用的模板以制得相對大量的納米線。眾所周知,在臨界膠束濃度(LMC)時,表面活性劑分子可自發(fā)形成圓柱形膠束[30](如圖 6a)。這些各向異性中間相結(jié)構(gòu)可用作為軟模板,伴隨著適宜的化學或電化學反應以促進納米棒的形成(如圖 6b)。有選擇的去除表面活性劑,以收集到所要納米棒或納米線(如圖6c)。基于這種原則,采用已知濃度的表面活性劑[31~32]如Na-AOT,Triton X、CTAB 等已制得CuS 、CuSe、CdS 、ZnS 、ZnSe等納米線。以晶種為媒介的生長過程也可合成高產(chǎn)量長徑比高達200的Au納米管[33]。首先加入檸檬酸,這涉及到Au納米顆粒的形成,接著依次加入十六烷基三甲基溴化銨(CTAB)、HAuCl4、抗壞血酸以及NaOH,通過反應即可制得。圖(5c) 為Au納米管TEM圖。
1.2.3 溶液-液-固過程(Solution-liquid-solid process) Buhro等[34]提出一種溶液-液-固過程(SLS法)合成Ⅲ-Ⅴ半導體[35]納米線晶體。在一個典型的程序中,低共熔點的金屬(In,Sn,Bi)可作為催化劑,通過有機金屬前驅(qū)體的熱分解反應制得所要的材料。通過SLS過程獲得的產(chǎn)品一般為單晶須。Korge等[36]采用超臨界流體-液-固過程(SFLS 法)合成大量的無晶體缺陷的Ge、Si納米線。
1.2.4 溶劑熱合成 溶劑熱法[37~38]以有機溶劑代替水,由于有機溶劑的多樣性,且具有較低的沸點和各異的介電常數(shù)、極性、粘度等,因此可根據(jù)不同的溶劑體系和目標產(chǎn)物設計出新的合成路線,從而擴大了水熱法的應用范圍。在此過程中,將溶劑與金屬前驅(qū)體和晶體生長調(diào)節(jié)劑或模板化試劑(如氨)的混合液置于高壓釜中維持較高的溫度或壓力以促進晶體生長和組裝過程。
筆者通過乙二胺溶劑熱反應制備屬立方晶系ZnS前驅(qū)體[43](ZnS?2en,en=乙二胺),然后在惰性氣氛中加熱該前驅(qū)體至一定的溫度熱分解得到單相六方晶系纖鋅礦型ZnS納米晶,二者均為納米棒構(gòu)型,Zn(en)2S的棒直徑約為30nm,ZnS的棒直徑較大,在60nm左右。
2前景與展望
以上主要總結(jié)了近幾年來常用的化學合成包括元素、氧化物、氮化物、碳化物、硫族化合物在內(nèi)的各族無機納米線的方法,并對其生長機理、結(jié)構(gòu)進行了探討。目前,合成高質(zhì)量、尺寸可控的無機納米線這一研究領域還相當活躍。毫無疑問,可以預見將會有更多的無機納米線被合成及表征,新的材料合成方法也會不斷的涌現(xiàn),化學方法在制備一維無機納米材料上將有著更為廣闊的前景。但目前各類方法多是經(jīng)驗總結(jié),并在此基礎上推測出部分反應機理,如能對反應機理進行更深入的研究,并找到普遍適用的方法,那將會極大的推動化學合成無機納米線方面的發(fā)展。
參考文獻
[1] S Iijima. Nature, 1991, 354: 56~58.
[2] T T Albrecht, J Schotter, G A K?stle. Science, 2000, 290: 2126~2129.
[3] M P Zach, K H Ng, R M Penner. Science, 2000, 290: 2120~2123.
[4] M H Huang, S Mao, H Feick et al. Science, 2001, 292:1897.
[5] Z W Pan, Z R Dai, Z L Wang. Science, 2001, 291: 1947~1949.
[6] Y Huang, X Duan, Q Q Wei. Science, 2001, 291: 630~633.
[7] Y Cui, C M Lieber. Science, 2001, 291: 851~853.
[8] Y Huang, X Duan, Y Cui. Science, 2001, 294: 1313~1317.
[9] H Vincent, F Chassagneux, C Vincent et al. J. Mat. Sci. Eng. A, 2003, 340: 181~192.
[10] T J Trentkr, K N Hickman, S C Goel. Science, 1995, 270: 1791.
[11] Y Wu, P Yang. J. Am. Chem. Soc., 2001, 123: 3165~3166.
[12] D Wang, H Dai. Angew. Chem. Int. Ed., 2002, 114:4977~4980.
[13] W Shi, Y Zheng, H Peng et al. J. Am. Chem. Soc., 2000, 83:3228~3230.
[14] C H Liang, G W Meng, L D Zhang et al. Chem. Phys. Lett., 2000, 329: 323~328.
[15] S T Lee, N Wang, Y F Zhang et al. MRS Bull. 1999: 36~42.
[16] N Wang, Y H Tang, Y F Zhang et al. Phys. Rev B., 1998, 58: R16024~16026.
[17] G Gundiah, F L Deepak, A Govindaraj et al. Topics in Cat., 2003, 24: 137~46.
[18] H Y Peng, X T Zhou, N Wang et al. Chem. Phys. Lett., 2000, 327:263~270.
[19] F L Deepak, C P Vinod, K Mukhopadhyay et al. Chem. Phys. Lett., 2002, 353: 345~352.
[20] F L Deepak, G Gundiah, A Govindaraj et al. Bull Polish. Acad. Sci., 2002, 50: 165~174.
[21] C N R Rao, F L Deepak, A Govindaraj, J. Mater. Chem., 2004, 14(4): 440~450.
[22] B Gates, B Mayers, B Cattle et al. Adv. Funct. Mater., 2002, 12: 219~227.
[23] B Mayers, Y Xia. J. Mater. Chem., 2002, 12: 1875~1881.
[24] B Messer, J H Song, M Huang et al. Adv. Mater., 2000, 12:1 526~1528;
[25] J Song, B Messer, Y Wu et al. J. Am. Chem. Soc., 2001, 123: 9714~9715.
[26] B Gates, B Mayers, B Cattle et al. Adv. Funct. Mater., 2002, 12: 219~227
[27] M Zheng, L Zhang, X Zhang et al. Chem. Phys. Lett., 2001, 334: 298~302.
[28] M J Zheng, L D Zhang, G H Li et al. Appl Phys Lett., 2001, 79: 839~841.
[29] M Pender, L Sneddon. Chem. Mater., 2000, 12: 280~283.
[30] Y Xia, P Yang, Y Sun et al. Adv. Mater., 2003, 15: 353~389.
[31] C N R Rao, A Govindaraj, F L Deepak et al. Appl. Phys. Lett., 2001, 78: 1853~1855.
[32] A Govindaraj, F L Deepak, N A Gunari et al. Israel J. Chem., 2001, 41: 23~30.
[33] B D Busbee, S O Obare, C J Murphy. Adv. Mater., 2003, 15: 414~416.
[34] T J Trentler, K M Hickman, S C Geol et al. Science, 1995, 270: 1791~1794.
[35] P D Markowitz, M P Zach, P C Gibbons et al. J. Am. Chem. Soc., 2001, 123: 4502~4511.
[36] J D Holmes, K P Johnston, R C Doty et al. Science, 2000, 287: 1471~1473.
[37] J Li, Z Chen, R J Wang et al. Coordination Chemistry Reviews, 1999, 190: 707~735.
[38] G Q Li, H Z Zhong, Z Chen et al. Semiconductor Photonics and Technology, 2004, 10(4): 256~259.
統(tǒng)計數(shù)據(jù)
共收錄化學品數(shù)據(jù)
147579 條
公告
更多>
-
微信掃一掃
關注:物競化學品數(shù)據(jù)庫
-
物竟數(shù)據(jù)庫 手機版
國內(nèi)首款化工類專業(yè)手機應用
-
微博賬號
wjhxp
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數(shù)據(jù)庫是一個全面、專業(yè)、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構(gòu)、檢測機構(gòu)、化學品工作者提供專業(yè)的化學品平臺進行交流。
數(shù)據(jù)庫采用全中文化服務,完全突破了中英文在化學物質(zhì)命名、化學品俗名、學名等方面的差異,所提供的數(shù)據(jù)全部中文化,更方便國內(nèi)從事化學、化工、材料、生物、環(huán)境等化學相關行業(yè)的工作人員查詢使用。
關注我們
-
微信賬號:物競化學品數(shù)據(jù)庫

-
微博賬號:wjhxp
聯(lián)系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
Copyright ? 物競數(shù)據(jù)庫 2009-2026.All Rights Reserved. 關于數(shù)據(jù)庫 | 關于物競 | 法律聲明 | 熱門關鍵字
 滬公網(wǎng)安備 31010602001115號
滬公網(wǎng)安備 31010602001115號