糖類在生物活性化合物合成中的應用
- 2012-12-24
- 專題
1. 介紹
在大學的實驗室里面,根據研究方向人們會給出一個獨特的稱謂,比如說“金屬催化劑專家”、“天然產品專家” 或者是沒有名字。我們的實驗室可以被稱為“糖專家”。
通常,對于化學家們來說糖類相當于于一個“強大”的工程。他們誤認為實驗室處理糖類必須需要專門的技術。然而,如果了解了糖的屬性,就可以用有機化學里面的常用方法。我們的實驗室目前從事糖化學-化學工藝兩個研究領域。雖然這兩個領域看似不同,但是它們共享基礎化學原則,并且彼此密切相關。從這個角度出發, 我們申請了以糖類化學為基礎的合成化學用以處理各種項目。
在這片文章中,將介紹我們最近的研究,從糖類合成生物活性化合物以及它們在郵寄合成化學和藥物化學中的應用。如果讀者通過這篇文章有機會更加熟悉了解糖類我們將倍感榮幸。
2. 糖化學
各種生物大分子將生命系統大致分為:蛋白質、脂類、多糖和核酸,它們分別相應的由氨基酸、脂肪酸、單糖和核苷酸構成。糖類的重要性通過兩個單糖和含有D-核糖的核苷酸作為他們的單元結構來進行證實。碳水化合物化學從單糖到多糖范圍廣泛。重要的是我們能從單糖的物理應用和化學特性中了解更多復雜的多糖。例如,糖化學包括單糖立體化學(微觀角度) 和宏觀角度上物理分析的多糖結構,比如,細胞壁上的肽聚糖。糖類化學因此與生物活性密切相關,并在同一時間延伸到所有合成化學領域。下一節我們將討論幾個重要的方面,糖化學,尤其是使用糖類合成化學。
糖類含有多種官能團,比如,羥基和氨基基團鏈接在他們的基礎骨干上形成手性化合物。由于這種手性特征,糖類可以具有立體化學復雜結構。這樣的化合物可以在溫和的條件下進行反應所以用于各種化學品中。例如,反應可以在水合條件下或者是完全中性條件下優先發生。這種限制可能似乎阻礙了實驗。但是從我們的角度來看,這意味著糖類合成方法真正適用于幾乎所有的其他化學品。這些都是嚴格意義上的“合成”。
如果我們了解了糖類的這些特點,那么我們就能夠從中獲取更多收益。例如,其廉價且易獲得,糖類常用于手性天然化合物的合成。1)糖鏈以及衍生物作為生物活性化合物的一個很重要的角色就是預期用作治療藥物將受到的關注。研究人員通過基元反應合成糖鏈或者是具有潛在生物活性的各種化合物參與到真正有用的化學中來。下一節將介紹我們合成唐衍生生物活性化合物的方法,我們將充分的體會到有機合成化學的本質。
3. 光學活性合成方法的發展,多取代環己烷及合成生物活性化合物的應用
例如D-糖類, D-葡萄糖為代表, 價格便宜并且制備容易,有效地利用這些化合物來合成光學活性化合物在有機合成和將來開發治療藥物中是一個不可或缺的任務。我們也別關注了多取代環己烷這個試劑在糖類生物活性化合物的全合成中的作用。
3.1 氯化鈀環轉換反應合成糖類的發展2)
光學活性多取代環己烯的合成方法已有很多的報道。尤其是自從Ferrier1979年利用汞化合物3)的[Ferrier (II)反應]研發的一個合成糖類的環化反應, 五元和六元環使用糖類轉換已被廣泛研究。4) 因為生物活性化合物往往天然存在一個具有多個官能團的環結構,這些環的手性中心立體結構的控制在這些化合物的全合成中變得至關重要。達到這一要求,再怎樣強調Ferrier (II)反應在具有復雜構想的化合物中的全合成中的重要性也不為過。5)
Ferrier (II)反應通過水溶液中汞化化學計量數據將5-烯橋吡喃糖苷轉換為環己酮(圖1)。
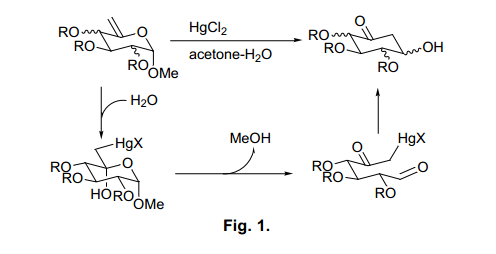
這是一個環的轉化反應,從四氫呋喃環島環己烷的轉換,反應步驟開始于烯烴的羥汞化,接著不穩定的半縮醛脫醇與開環反應,二酮分子內羥醛縮合。由于從糖類生物合成肌醇的反應使用類似反應機理,6)這個反應得到越來越多的關注。因此,Ferrier (II)反應在它的反應機理中產生了一個有趣的現象。然而同時在使用汞化反應是有一個嚴重的問題。盡管汞化催化量最近由Lukacs等7)和Ogawa等8)的報道已經足夠充分,但是從對環境和人類健康的考慮出發,開發更為實用的方法應以滿足目前的需求是很有必要的。因此,我們使用其他的金屬鹽來分析Ferrier (II)反應。
我們側重于反應的第一步,烯烴中加入水,類似汞一樣搜索催化含氧金屬化的過渡金屬。結果,我們發現二價鈀具有良好的活性,尤其是,氯化鈀的催化活性和三幅乙酸汞一樣高。Adam 1988年報道過Ferrier (II)反應中氯化鈀的應用9) ,但是對其一般的應用沒有討論。10) 然而,因為這個反應實在中性溶劑中進行,所以沒有副反應比如說含氧官能團的β-消除,或者是阻塞基團的消除。此外, 氯化鈀易于處理。基于這些方面,我們選擇氯化鈀作為Ferrier (II)反應催化劑。
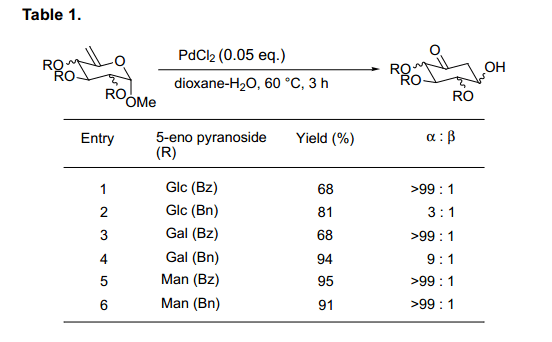
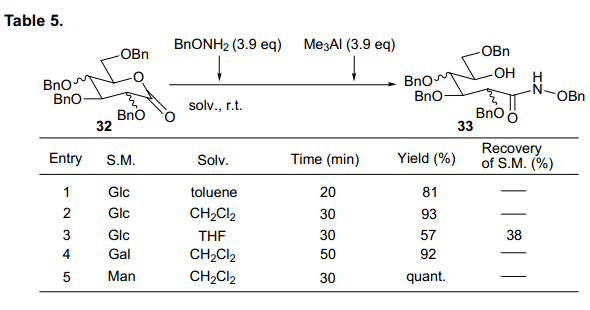
雖然最初氯化鈀的穩定性是一個問題,我們發現這個化合物在水合二惡烷中保持穩定。如圖表1所示, 從葡萄糖、半乳糖和甘露糖而來的各種基質衍生物,與0.05當量的氯化鈀反應轉換為環己酮,產品收益率高。
由葡萄糖和半乳糖的來的5-烯吡喃糖苷表明了在苯甲酰基和芐基形式之間有反應性差異。此外,我們發現新形成的羥基基團立體化學選擇性與每一個基質相隨 (條目1-4)。相反,甘露起源的基質在保護集團中不顯示任何的差異,與α-選擇性得到相應的環己酮(條目5, 6)。
環完全轉換通過(0.05 equivalent)當量的氯化鈀催化劑。這個反應比以往的轉換方法更具優勢,因為:1)它非常普遍并且適用于各種糖類的環轉換,2)反應在非常溫和的條件下進行,3)反應中通過不同的立體化學選擇性產生的各種異構體來源于不同于汞化反應的同一種化學機理 。此外,因為氯化鈀催化劑和溶劑都不需要提純,所以這個反應實用,并且可以應用到工業生產中。利用這些優點,我們產用這個方法來完成生物活性化合物的全合成。
3.2 Cyclophellitol11)的全合成
Cyclophellitol 是一個由Umezawa等人在1990年分離出來的一個β-D-葡萄糖苷酶抑制劑。12)近年來, 人們發現糖相關酶與細胞內識別密切相關,13)這些酶抑制劑一直被用來重點研究各種疾病的治療藥物方面。14 )特別是,Cyclophellitol具有很高的活性,它的應用有望延伸到制作防病毒和抗-HIV劑抑制癌細胞的擴散。15)Cyclophellitol的結構是一個在葡糖糖構像多取代環醇β –位置上類似β-D-葡糖苷的環氧環。16) Cyclophellitol合成方法已經被廣泛應用到世界各地。17)我們不僅僅研究cyclophellitol也包括它的差向異構體的合成方法,最終的目的是構效關系的研究。
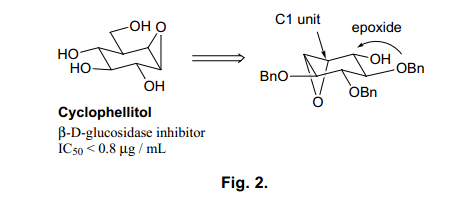
Fig. 2顯示了cyclophellitol和它的差向異構體的合成方案。在Ferrier (II)反應中,糖類用作初始基料轉化為環己烷環,然后形成一個立體選擇性環氧環。然后利用區域選擇性和立體選擇性親核加成環氧環,一個羥甲基基團被引入如圖C1位置。最后,進行消除反應形成一個β-構型環氧環來完成cyclophellitol的全合成, 其中所有的取代基都由立體化學來控制。這條線路可以允許cyclophellitol多種差向異構體的合成。
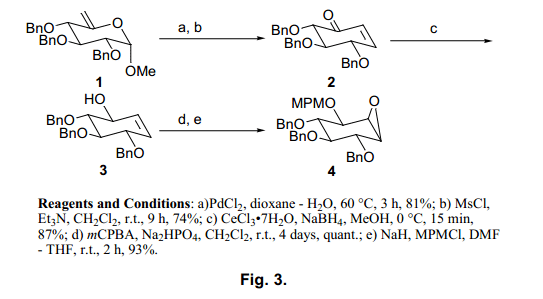
在我們的試驗中,5-烯醇吡喃葡糖苷118)在Ferrier (II)反應通過常規方法首先得到(利用氯化鈀催化劑得到環己酮)。環己酮通過消除反應轉換為烯酮2。烯酮2在Luche’s條件19)下還原得到β-醇3。然后α-型環氧物立體選擇性構象形成, MPM基團保護羥基得到中間體環氧化物4(圖3).
在全合成途徑中關鍵反應是區域選擇性和親核加成進攻環氧化物4羥甲基基團。一般情況下,環己烷的開環反應中親核試劑主要進攻環氧化物的軸向位置。20)因此, 我們預計親核取代環氧化物將在軸向位置,C5處,并且不顯示所需的區域選擇性21) (圖4).
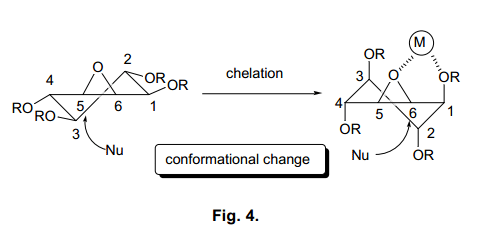
我們設想如果我們能夠改變環氧化物的構象,那么我們就能夠在C6位置引入羥甲基基團 。
換言之,如圖4所示, 環氧化物中金屬和氧原子之間的螯合和醚可能直接改變環己烷環的構象,從而導致軸向親核進攻的位置在C6處,而不是C5處。對于這樣的,我們使用一個硼酸試劑來取代羥甲基,來研究環氧化物使用基料在被保護位置C1(利用各種保護劑,同位進攻)區域選擇性(表2)。22)
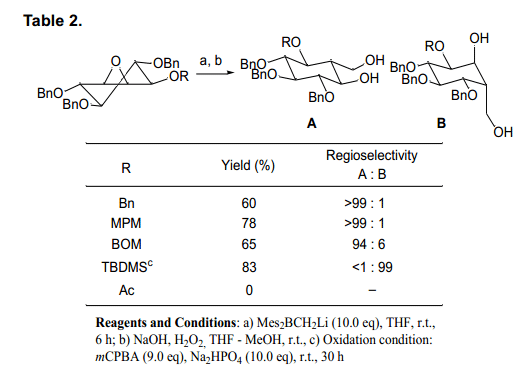
遺憾的是,由于酰基保護基團和硼酸試劑反應,我們沒有得到羥甲基的加成產物。然而,羥甲基與基板上的保護基團醚以很高的速率發生反應。非常有趣的是,基質由芐基保護,在相反的位置由于螯合作用MPM 和BOM基團生成羥甲基產品A,但是保護基團是TBDMS 時生成B。我們假設巨大的TBDMS基團可能會阻止醚氧原子,這樣的結果,那么環己烷結構不會發生轉換。基于這些結果,我們選擇了MPM 作為做為最合適的保護劑,并進行了如下圖所示的合成(圖5)。
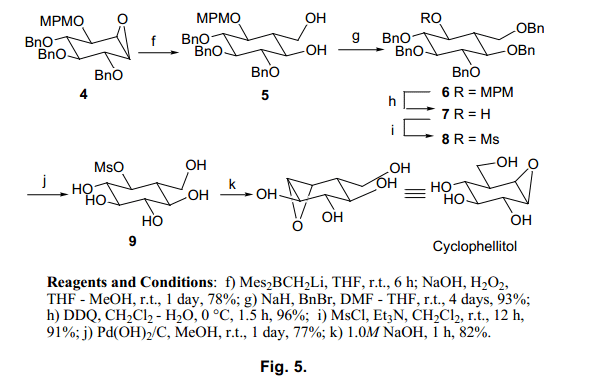
羥甲基化后得到二醇5,它由芐基基團保護,并且甲基與一個MPM基團進行交換。然后,所有額芐基通過催化氫化脫保護得到五醇9。五醇在堿性條件下很容易的進行環氧化物環化反應,Cyclophellitol 從原料1最終合成,總收益率在14%。
在這個合成方法中,初始糖類很容易的導致各中差向異構體,這取決于糖類的構象和保護基的類型。同樣的方法,C3處一個不同的排列,可以成功的得到Cyclophellitol的一個差向異構體。23)
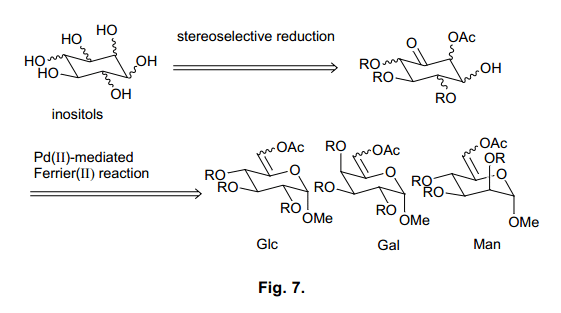
3.3 肌醇異構體的合成24)
肌醇是動物和植物中都能發現的生物活性化合物之一,在細胞的增值和癌變中表現出多種功能。25)這里共有九中類型的肌醇立體異構體(圖6), 因為在細胞內信號轉導途徑研究的最新進展,myo-肌醇得到了廣泛的關注。26,27)
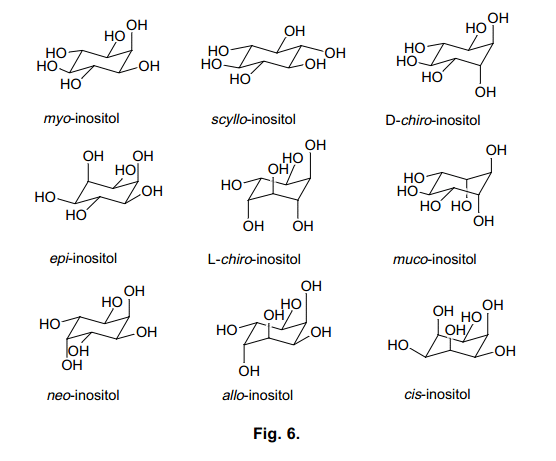
例如, 細胞外刺激反應,myo-肌醇-1,4,5-三磷酸[Ins(1,4,5)P3]調動Ca2+濃度增加。28)此外,myo-肌醇-1,3,4,5-四磷酸[Ins(1,3,4,5)P4]從細胞外圍中接收Ca離子。29)由于肌醇多聚磷酸鹽通過這種機制通過改變細胞內Ca離子濃度發揮不同的功能,它們被稱為第二信使。盡管更多的myo-肌醇多聚磷酸鹽被發現,但是由于他們的濃度低,難以隔離,它們的激活機制還是難以琢磨。30) 因此,得到肌醇衍生物作為肌醇受體的配體十分必要。31) 然而,只有四種天然存在的肌醇異構體(scyllo-,neo-, D-手性-, L-手性-肌醇)可以作為myo-肌醇類的激動劑或者是拮抗劑,其他的四種(cis-,allo-,epi-和muco-肌醇)只能通過化學合成獲得。其中,有兩種類型容易獲得,但是它們非常的昂貴。這完全歸因于肌醇異構體罕見的結構和合成方法的不實際性。為了建立一個簡易的合成方法,來合成這九種類型的肌醇異構體包括其中我們之前沒有關注過的類型,我們設計了一個合成途徑如圖7所示。.
這條線路,我們預期含有一個乙酰氧基的5-烯吡喃糖苷通過Ferrier (II)反應在C6位置江北轉換為環己酮,環己酮中的酮部分被還原產生一個肌醇骨干。32)利用從葡萄糖、半乳糖、甘露糖衍生得到的基料,這個方法在同時間內將會產生種類繁多的異構體。
3.3.1 6-O-乙酰基-5-烯吡喃糖苷的Ferrier(II)反應審核
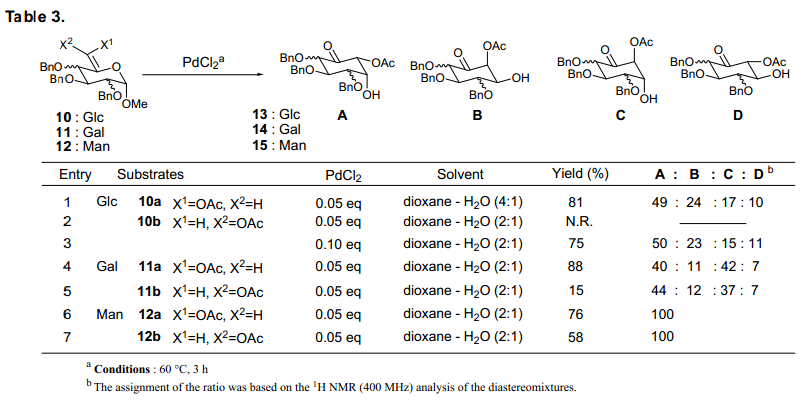
首先,Ferrier (II)反應6-O-乙酰基-5-烯吡喃糖苷與基料反應,用實驗當量的氯化鈀做催化劑進行檢測。我們尤其關注烯醇酯在立體化學方面的影響,對E-型在反應性和立體選擇性上面進行了比較(表3)。
葡糖糖衍生物基料10 Z-型比其E-型的反應性高,并且生成了四個對應立體郵購提混合物。D-半乳糖起始物11也出現了類似情況。對于每一個糖類,從Z-型或者是E-型得到的四核環己酮的比率在C6構象上是沒有任何影響的。當使用甘露糖衍生物基料12時,Z-和E–型都得到一個單一的環己酮構象。這些結果的基礎上,我們發現任何糖類都有效地進行著環轉換。我們還發現根據原始糖類的構象,環己酮形成的比率不同,基料上C6位置的構象不會保留,并且C6不會影響新的手性中心構象。33)我們也分析了反應中使用Hg++, 但是慢于氯化鈀催化反應,并產生不同比率的異構體。因此,我們得出結論,氯化鈀竟會帶來一個令人滿意的效果,并進行下一步的討論。
3.3.2 立體選擇性還原反應的審核
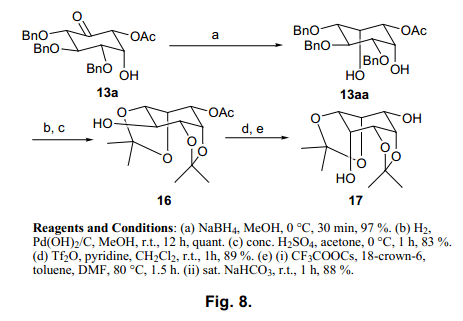
我們測試了兩種還原方法得到環己酮(表4)。當使用Me4NHB(OAc)3時34),除了和其他的反應,在14c (條目11)中不能繼續進行,生成的醇在羰基β–位置處被反式方式還原。尤其是與13a、13c、14a、15a 反應立體選擇性非常高,僅得到β-型,產率很高。我們認為是羰基β–位置處的羥基還原的原因。另一方面,當使用硼氫化鈉時,反應在空間位阻小的位置進行,13a, 14a, 14b, 14c和15a高選擇性的還原產物。因為這兩個還原反應彼此互補,所以通過改變反應可以輕松得到所需醇。因此,這個立體選擇性的方法讓我們得到了所預期的九分之八的肌醇。最后一個異構體,cis-肌醇通過13a獲得(通過葡萄糖衍生物的環轉換,產率很高)。

如圖8所示,13a中的酮通過硼氫化鈉還原只得到α-型醇13aa,產率很高。然后,芐基脫保護,縮丙酮基團用于選擇性保護cis-二醇。最后一個羥基集團反轉得到cis-肌醇衍生物17。這些肌醇衍生物脫保護,被證實了,與先前報道的天然存在的衍生物具有相同的值。36)
因此,我們成功的立體選擇性合成了肌醇的所有立體異構體。該方法使用利用氯化鈀作為催化劑的Ferrier (II)反應制得的多種環己酮異構體,能夠同時合成多種異構體,這是使用汞化鹽所得不到的。
3.4 肌醇多聚磷酸鹽的合成
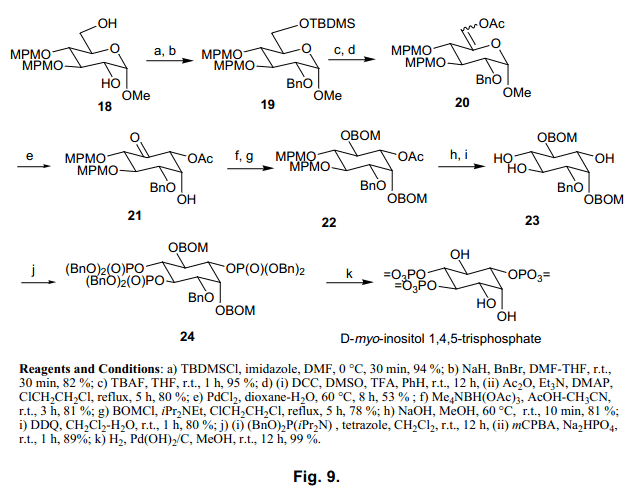
接下來,我們合成Ins(1,4,5)P3和Ins(1,3,4,5)P4, 如圖9、10.所示。用這個方法得到的Ins(1,4,5)P3和Ins(1,3,4,5)P4 顯示出來的值與天然存在的化合物一致,37,38) 我們對其結構也進行了確認。
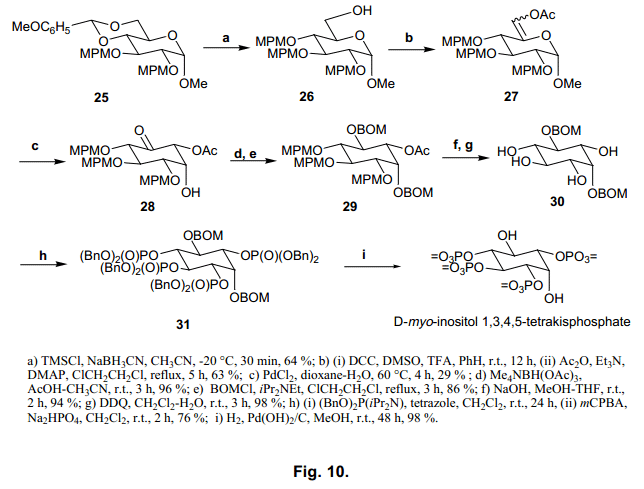
接下來的部分,我們將描述內酯糖對合成各種生物活性化合物和糖鏈的有效應用。
4.葡萄糖-D-內酯合成L-糖的應用39)
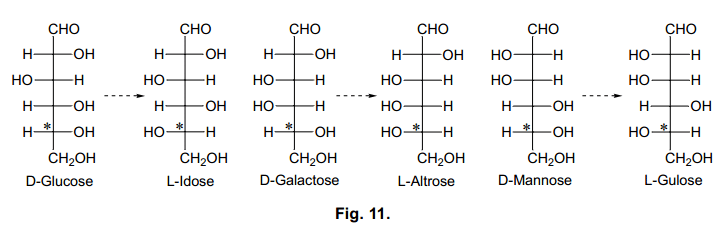
自然界中L-糖少有產生,但有趣的事,在生物活性化合物的活性方面它往往扮演者一個關鍵的角色。40)然而,它很稀有,并且合成方法尚未建立,L-糖的研究遠遠落后于D-糖。為了克服這種情況,使得L-糖像D-糖一樣得到發展,我們制定目標設計制備L-糖的實用方法。 為了接近化學合成,我們試圖在C5位置處有效地將D-型轉換為L-型。
如圖11所示,D-葡萄糖在C5處轉換為L-艾杜糖,D-半乳糖轉換為L-阿卓糖。盡管L-艾杜糖和L-古洛糖都是有市售的,但是它們非常的昂貴。L-阿卓糖沒有銷售市場。我們的方法是利用廉價易得到的D-糖通過簡單的步驟來有效地合成L-糖。我們試圖通過甘油-D-內酯羥肟衍生物分子內環化和合成L-糖(Mitsunobu反應),這是一個D-糖在C5位置典型的SN2-型反應。
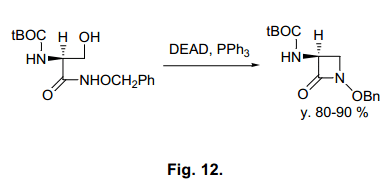
Miller等人已經報道了氨基酸β-羥基異羥肟衍生物的分子內環化反應。41)這個反應已經廣泛應用在各種β-內酰胺抗生素的合成上面,因為親核進攻酰胺氮原子主要生成β-內酰胺(圖12)。然而,不僅僅是酰胺氮原子,還有羰基氧也會進攻化合物。事實上,其他的基板可能會產生內酯,42) 我們感興趣的是糖衍生物基板被使用過后的結果(圖13).
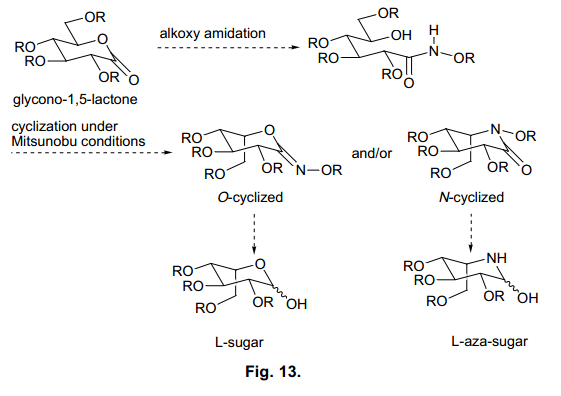
如圖13所示,親核進攻酰胺N原子生成N-環化物,親核進攻氧原子得到O-環化物。在這種情況下,Mitsunobu反應在C5反轉生成環化物;N-環化物得到L-氮雜糖, O-環化物生成L-糖。基于這一步驟,我們分析了D-glycono-1,5-lactones的δ –羥基烷草酰胺衍生物的分子內環化。
基板δ-羥基異羥肟衍生物通過D-葡萄糖吡喃內酯、D-半乳糖吡喃內酯、D-甘露糖酸吡喃內酯與芐胺反應合成。我們預測該反應可由路易斯酸促進反應,并且發現三甲基鋁對高產率合成δ-羥基異羥肟衍生物起到一個顯著的加速效果,如圖5所示。43)同樣的,烷氧基鹽酸鹽證明了在有效合成δ-羥基異羥肟衍生物時顯示出很高的產率。根據這些,我們利用Mitsunobu 反應分析了化合物的環化反應。
環化進程迅速,得到O-環化和N-環化δ-羥基異羥肟衍生物,產率分別為71%和13%,O-環化是N-環化產率的5.5 倍。這與Miller等人在β-內酯的獲得結果上不相同。D-半乳糖衍生基質也主要生成O-環形式。有趣的是,D-甘露糖主要產生O-環化物而不是N-環化物。基于這樣的結果,我們發現糖類δ-羥基異羥肟衍生物主要優先生成O-環型,盡管從O-型到 N-型環化物的產率比隨著初始糖的類型而改變。此外,溶劑影響的詳細分析表明,O-型到N-型環化物的產率比隨著使用溶劑D-葡萄糖和D-半乳糖的溶劑類型改變而變化。這個環化反應的簡易機制如圖所示(圖14).
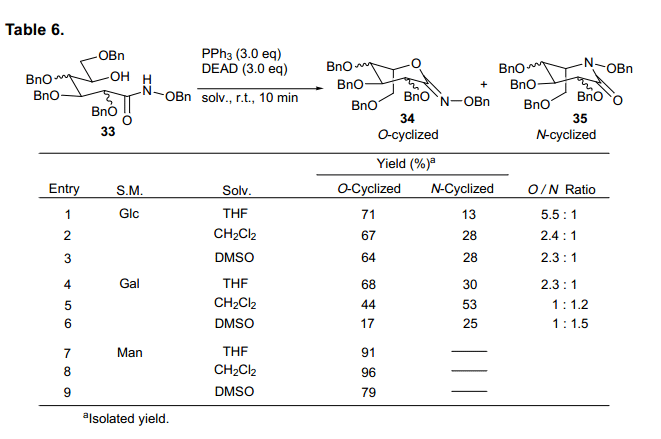
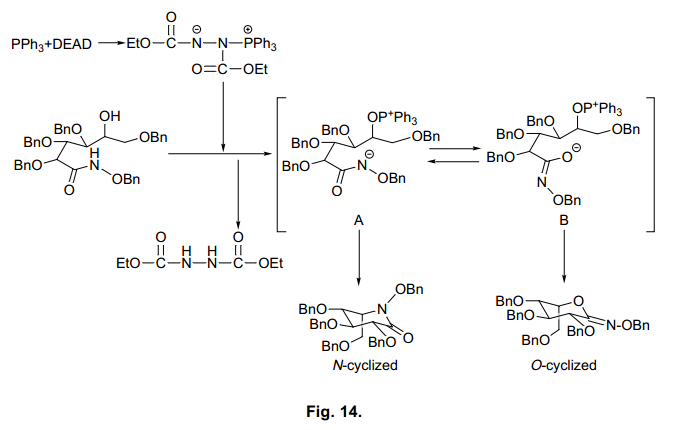
首先,復合物控制通過Mitsunobu試劑使得δ-羥基異羥肟衍生物的酰胺氫生成中間體A ,其氮原子上含有一個負離子電荷。在C5處親核進攻氮原子生成N-環化產品。另一方面, 中間體A通過負電荷轉移到氧原子上面生成中間體B。親核進攻氧原子生成O-環化產品。目前為止原因尚不清楚,但是當使用δ-羥基異羥肟衍生物時,通過中間體B形式的環化反應占主導。
如果我們使用任何糖類合成L-吡喃糖,優先得到的是O-環化型(圖15)。
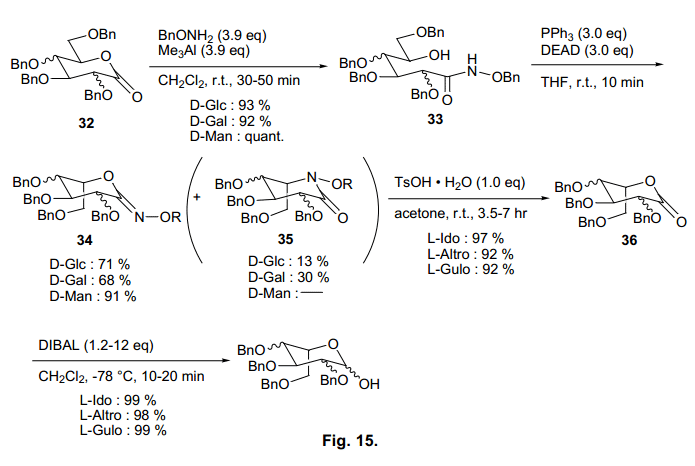
由于O-環化品34(這三種糖衍生物)在L-吡喃丙酮C1處脫保護, 所以我們在酸性條件下脫保護分別制得L-idonolactone衍生物,L-altronolactone衍生物和 L-gulonolactone衍生物,產率很高。然后, C1位置羰基被DIBAL還原,分別有效地獲得L-艾杜糖, L-阿卓糖和L-古洛糖衍生物。特別是,通過這四步,顯示得到83%的D-甘露糖,證明了這個方法的高效性。最終的產品L-吡喃衍生物僅在C1位置不受保護,因此可以進一步用于糖基化。因此,我們首次建立了從稀有L-糖制備D-糖的簡易廉價合成方法。
接下來,我們試圖通過此幻化方法,從L-核糖合成糖1,4-內酯衍生物(圖16)。L-核糖是D-核糖的對應異構體,自然界中少有產生。最近,L-核糖的合成方法得到了越來越多的發展,因為含有L-核糖的化合物顯示出各種生活活性包括病毒影響。44)
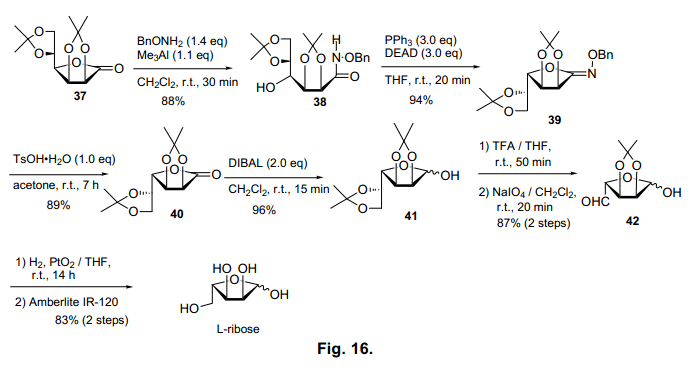
L-核糖的合成基于同樣的方案。內酯37由市售產品D-甘露糖-1,4-內酯常規方法衍生而得,環裂解后,通過Mitsunobu反應再次環化。在這個方法中,僅僅有O-環化物39生成(C4位置倒轉),沒有N-環化產品物。我們認為甘露糖衍生物1,5-內酯得不到上述的N-環化產物,我們得出的結論是,在這個方法里甘露糖只專門生成O-環化產物。雖然尚未不能明確的證明,但是糖C2 位置的羥基集團構象似乎影響O-和N-環化產物的比例。特別是, D-甘露糖-1,4-內酯有利于合成L核糖, O-環化物得到后,39在C6處被氧化切割,通過幾個步驟就可以有效地得到L-核糖。
如上所述,這個方法可以由N-環化物派生出氮雜糖(分子間環化反應的副產物),因此,糖相關酶的抑制劑可能應用到。
5. 利用糖原酸酯合成糖衍生物
糖原酸酯是通過糖內酯和糖二醇組合形成螺環而得。他們獨特的結構最初是在一系列orthosomycin抗生素中發現的。45)這種來自于糖苷鍵的獨特的特性鍵,用來研究rthosomycins的合成和結構分析,專注于臨位酯鍵形成的方法。其結構是,糖原酸酯本身的結構特性和反應性和他們的應用已經have been left unknown。然而,這個特定反應性的戰略應用對有效合成方法的進展和使用原酸酯作為糖固定結構似乎成為可能。從這一個角度來看,我們分析糖原酸酯的各種合成方法旨在表征這些化合物和他們的反應性。
5.1糖原酸酯的高效合成方法的發展46)
在我們剛開始的研究中,糖原酸酯的合成報道很少,并且有效合成糖原酸酯的數量也很少。因此我們試圖開發更為有效地方法。通過由Miyat等人分析縮酮的合成方法, 47)我們發現在過量的methyl trimethylsilyl ether (TMSOMe)和一定數量的trimethylsilyl triflate催化劑的存在下,糖原酸酯從糖內酯和二醇中有效地生成。利用甲苯在反應中減壓條件下提高產率,除去副產物甲醇和六甲基二硅氮烷,當使用相當量的內酯和二醇時生成各種糖原酸酯(圖17)。
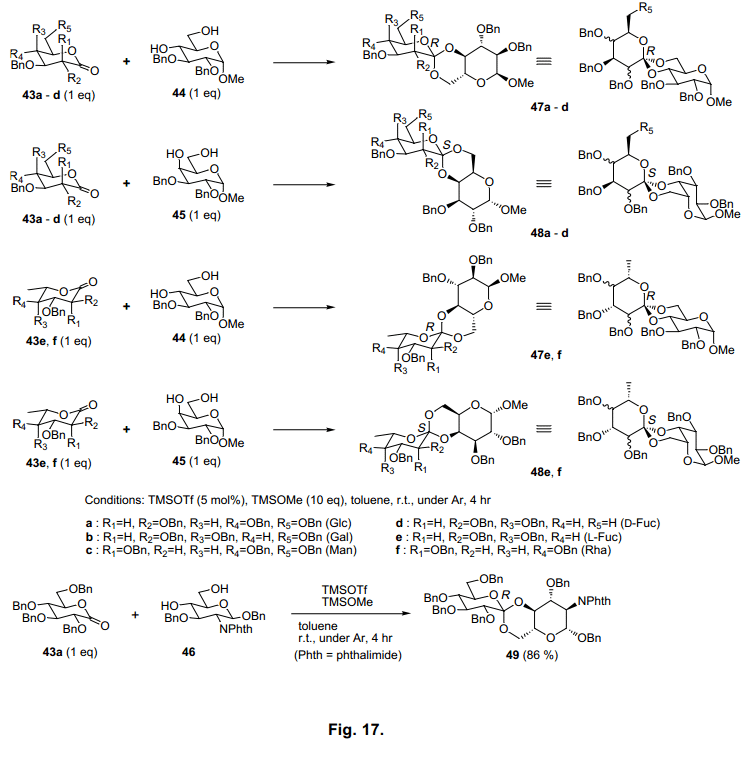
基于螺碳周圍的結構,糖原酸酯形成兩個異構體。這兩種異構體中的任何一個都會有選擇性的生成糖原酸酯(47a-f,48a-f, 49)。
5.2 糖原酸酯的結構分析48)
用X-ray 分析團原酸酯的兩種異構體的結構是必須的。我們使用電腦結合投射分析和構象分析分析了糖原酸酯的結構。
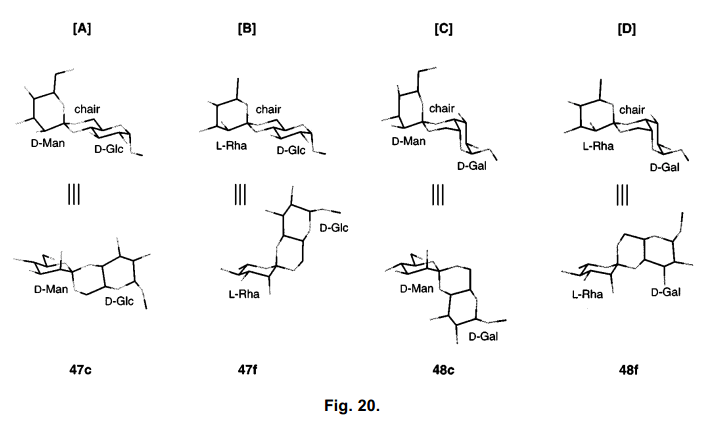
前一節中我們對鄰位酯和吡喃二惡烷的3-環骨架的描述,我們用X-射線分析了47c以及47f、48c、48f (50-52)等的乙酰衍生物的晶型,螺碳的構象被分別的確定為R、R、S、S。(圖18)

接下來,假定我們得到R和S-構象,我們估計通過使用使用LOMD方法50)由特定的力mm2*在MacroModel 6.049)得到的原酸酯的構象是最為穩定的。更為穩定的構象是通過計算每一個糖原酸酯的構象能量來確定的。在每一個化合物中,兩個異構體之間的能量差別足夠的大, 47a-f螺碳確定是R-構型,而48a-f確定為S-構型。這個結構與Yoshimura等人報道的數據一致。51)
糖原酸酯結構計算的分子透露了兩個構象能量差異源于它們環結構骨架的穩定性差異。圖19顯示47c的設定R-與S-異構體的環結構骨架。R-構型, 甘露糖吡喃環氧原子與中心手性二惡烷成軸向,估計會更加穩定,由于端基異構體的效果,這種構象被大大的優先選擇。52,53) 同樣的,其他的11種糖原酸酯,這些構象中的其中任何一個酯結構都比其他的要穩定。據推測,不利的異構體具有一個扭曲的船型構象二惡烷填補在吡喃環的赤道軸位置,如圖19所示。

這些結果表明,一定范圍內的羥基構象的差異,不影響吡喃環的結構,也不影響螺碳和優選結構骨干的構象。因此,合成的糖原酸酯可以分為四個結構基團:glycosyliden的D-或者是 L-型,二醇中的葡萄糖或者半乳糖。例如,圖20顯示從兩個方向觀測47c、47f、48c、48f的結構。此外,與47a相關的49的構象的分析,估計其具有A-型環結構。
5.3 糖原酸酯還原糖基化的發展54)
圍繞糖原酸酯結構分析的有機化學方法,其反應性鮮為人知。我們分析糖原酸酯與氫和甲基陰離子的反應性有望開發一個新的合成方法。圖21 顯示了陰離子與糖原酸酯反應的廣義方程式。
陰離子進攻,在a-位置裂解環生成縮醛或者是縮酮化合物,而在b-或者c-位置處裂解則生成糖苷。每一個產品在新形成的手性中心都有兩種構象。如果哦我們能控制區域選擇性開環和陰離子立體選擇性進攻,那么我們就可以研發出一個新的合成方法。盡管在研究糖基化發展中激活糖供體已經收到關注55),我們嘗試合成糖鏈更為靈活的方法。
當糖原酸酯47a被氫離子還原,會有四種可能的產物具有其中α(1→4),β(1→4),α(1→6)或β(1→6)鍵。當我們評估各種還原試劑的有效性、區域選擇性和立體選擇性的時候,發現ethermethylene chloride 中兩種等量的LiAlH4和AlCl356)選擇性從47a生成β(1→4)糖苷53a產率92%(表7, 條目1)。這種情況下,其他三種預測的糖苷沒有生成,表明了其高區域選擇性和立體選擇性反應。使用這個方法,糖原酸酯47b-d, 48e和f還原產率為92-99%,并選擇性生成β (1→4)糖苷53b-d, 54e和f (表7)。然而,其他的糖原酸酯47e, f和48a-d的還原不能有效的進行。進一步評估還原劑,我們發現氰基硼氫化鈉和氯化鋁在甲苯-乙腈57)中有選擇地產生β(1→6糖苷55a-d,56e和f(表8)。然而,當我們采用這種方法合成糖原酸酯47A-D,48E和F時,它沒有給出合適的產品。
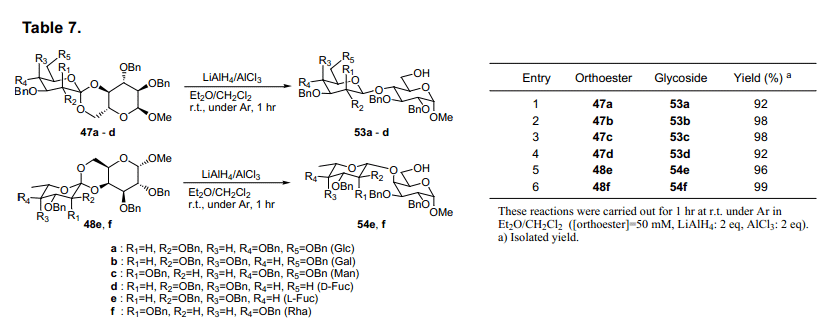
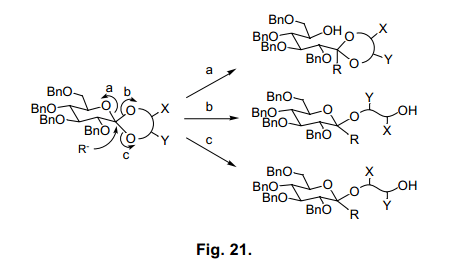
這些糖原酸酯中所發現的反應性差異可以通過上述構象差異來進行簡單的解釋。47a-d, 48e和f 骨干環結構預計為A或者D型,如圖20所示。圖的末端二醇環C6處氧原子與內酯吡喃環成軸向。另一方面,化合物47e, f和48a-d預計具有B或者C型,如圖20,但是C4氧是軸向。由于極端異構體的影響,我們推測吡喃環在軸向比在赤道位置具有更高的反應性58), 但是如果使用適當的還原劑,由軸向裂解而得的糖原酸酯會優先獲得。
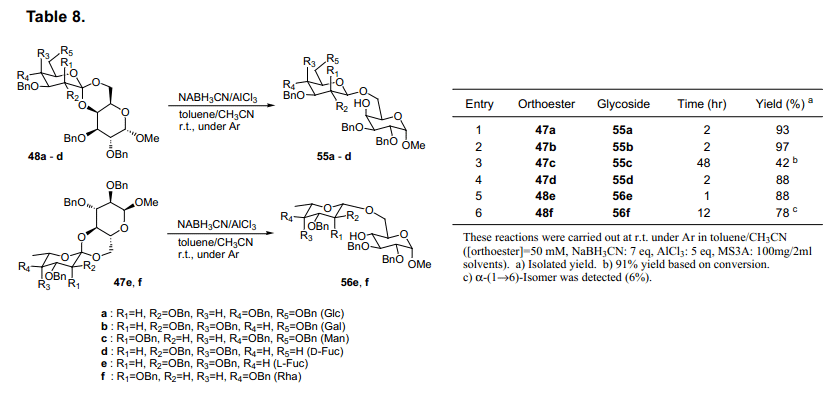
我們也進行了糖原酸酯57a-c通過使用還原試劑LiAlH4/AlCl3的還原反應。當57a-c主要形式使用時,預計軸向位置C3氧原子, 可以選擇性的還原生成β(1→4)糖苷58a-c,產率92-97% (表9)。
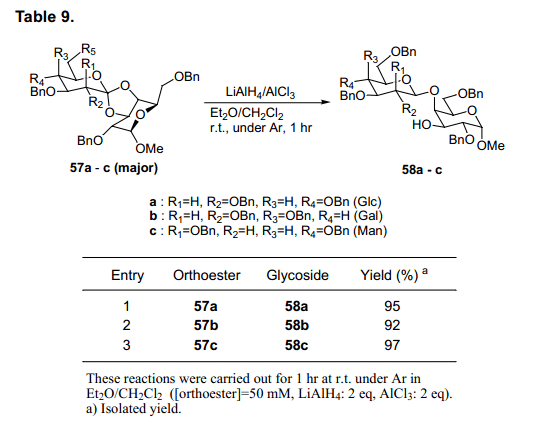
在這個還原條件下,57a-c的次要形式幾乎不進行反應。因此,當該產品不進行異構體分離就用于還原反應,那么生成的β(1→4)糖苷不具有任何實在問題。如圖22所示,鄰氨基葡萄糖酯也進行了分析。49轉換為二芐基保護形式60后,通過 LiAlH4/AlCl3還原反應選擇性生成β(1→4)糖苷61。不幸運的是,具有鄰苯二甲酰亞胺的葡萄糖胺49 或者不受保護的氨基基團59都不適合做反應的基料。
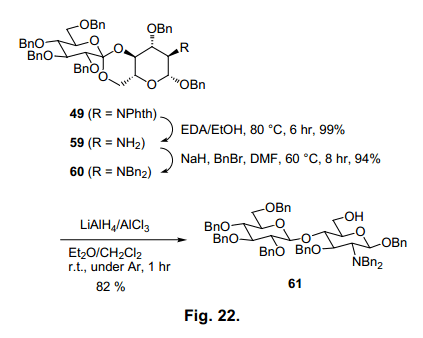
這個糖基化方法中最有趣的一方面就是β-甘露糖苷和β-鼠李糖苷的獲得都具有很高的選擇性。傳統的方法很難生成cis-1,2-β-糖苷,但是新的方案就允許β-糖苷從甘露糖或者鼠李糖中選擇性生成。原酸酯的形成和還原產率都是很有效的,并且等同或者高于其他合成cis-1,2-β–糖苷的方法。59-60)
6. 糖原酸酯合成卡巴糖方法的發展61)
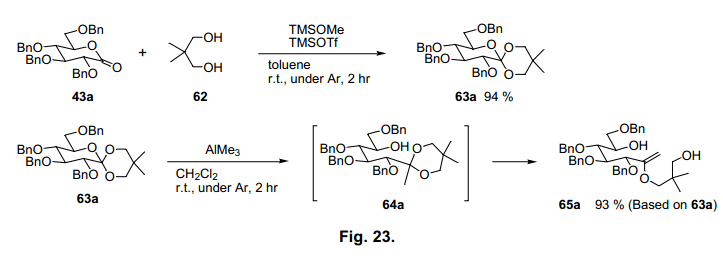
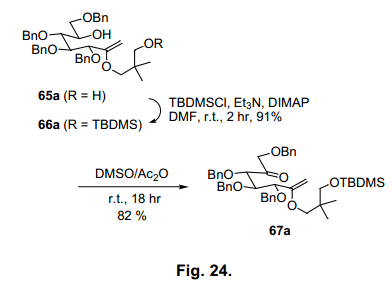
如前所述,合成環醇的各種方法已被廣泛的研究。我們開發了一個利用原酸酯和甲基陰離子反應,合成卡巴糖的一個簡單的合成方法。過量的 糖原酸酯63a和三甲基鋁反應,預計有一個甲基陰離子供體,得到烯醇醚化合物65a,產率93% (圖23),由于縮酮64a分離作為中間體,反應初始步驟必須通過引入甲基陰離子進行環裂解圖21所示。接著二惡烷環碳氧裂解62)伴隨三甲基鋁質子化 生成烯醇醚65a。烯醇醚65a具有七個碳原子,因此用作于卡巴糖的前體。我們試圖將65a 轉換為aketone 67a (圖24),通過羥醛縮合形成環己烷。63)
我們發現加熱兩種等量的氯化鋅在水合四氫呋喃64)可以將67a轉換為卡巴糖68a,產率90% (表10,條目5)。
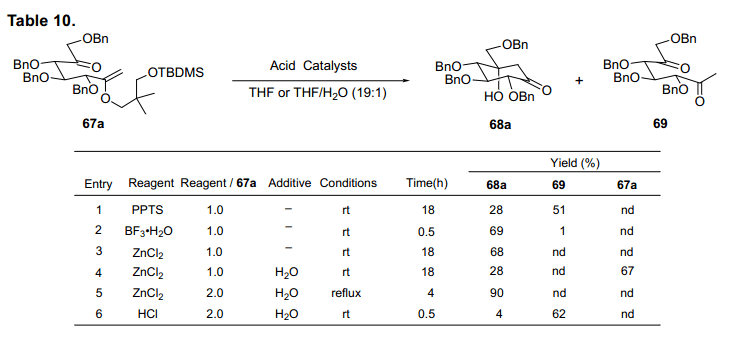
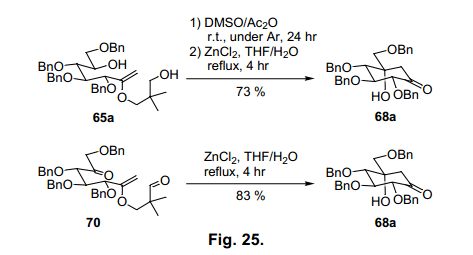
條目3中,當氯化鋅用在脫水溶劑中,反應加速但是有副產物生成,因此所需化合物產率降低。然而,少量水加入,就會生成高產率的68a(條目5)。此外,鹽酸加入水合tetrahydroflan,主要生成二酮69(條目6)。當我們省去甲硅烷基保護簡化程序使用烯醇醚65a 直接氧化的粗產物用于羥醛縮合反應,得到68a產率73%(圖25).
第一氧化反應主產物是二羰基70, 接著隔離和羥醛縮合反應后,酮選擇性轉換為68a (圖25)。同樣半乳糖43b和甘露糖43c內酯生成卡巴糖68b和c,產率分別為64%和56% (圖26)。
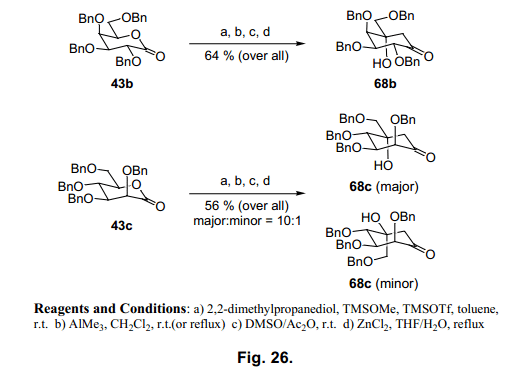
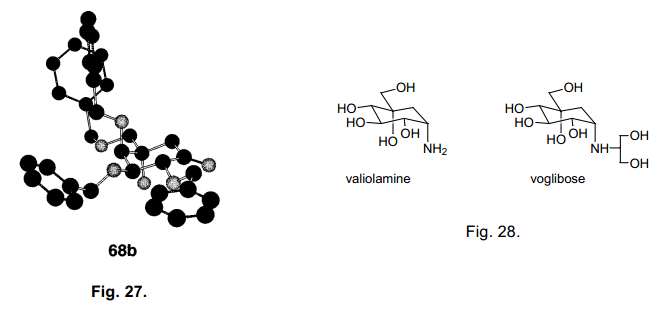
當使用68c時,羥醛縮合環化生成兩種異構體10:1。兩種異構體68b和68c構象通過X-射線分析圖27),通過環狀苯基硼衍生物的形成。65)68a 是一個合成valiolamines衍生物非常有用的synton,66-67) 我們也合成voglibose (圖28), 一個糖尿病藥物,根據Fukase的方法。68)
7. 結語
雖然本文中所述方法目標都是合成糖類,我們仍然通過這些方法以及新的合成方法關注通過常規反應的糖烯醇或內酯的合成。糖類是廉價并且易獲得的光學活性化合物資源。我們相信這些糖類的方案可以極大的促進醫藥化學和有機合成化學的發展。
References
1 Bols M., “Carbohydrate Building Blocks”, John Wiley & Sons, Inc., New York, 1996.
2 Iimori T., Takahashi H., Ikegami S., Tetrahedron Lett. , 37, 649 (1996).
3 a) Ferrier R. J., J. Chem.Soc., Perkin Trans.1, 1455 (1979). b) Ferrier R. J., Chem. Rev., 93, 2779 (1993).
4 Ito H., Motoki Y., Taguchi T., Hanazawa Y., J. Am. Chem. Soc., 11 5 , 8835 (1993).
5 Chida N., Ogawa S., Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 53, 858 (1995).
6 Wong Y-H. H., Sherman W. R., J. Biol. Chem., 261, 11083 (1985).
7 Mechado A. S., Olesker A., Lukacs G., Carbohydr. Res., 135, 231 (1985).
8 Chida N., Ohtsuka M., Ogura K., Ogawa S., Bull. Chem. Soc. Jpn. ,64, 2118 (1991).
9 Adam S., Tetrahedron Lett., 29, 6589 (1988).
10 The glucosamine-derived substrates are studied, but the yield is not high. László P., Dudon A., J. Carbohydr. Chem. , 11, 587(1992).
11 Takahashi H., Iimori T., Ikegami S., Tetrahedron Lett., 39, 6939 (1998).
12 a) Atsumi S., Umezawa K., Iinuma H., Naganawa H., Nakamura H., Iitaka Y., Takeuchi T., J. Antibiot., 43, 49 (1990). b) Atsumi S.,Iinuma H., Nosaka C., Umezawa K.,J. Antibiot.,43, 1579 (1990).
13 Sinnott M. L., Chem. Rev., 90, 1171 (1990).
14 a) Jespersen T. M., Dong W., Sierks M. R., Skrydstrup T., Lundt I., Bols M., Angew. Chem., Int. Ed. Engl., 33, 1778 (1994). b)Knapp S., Naughton A. B. J., Dhar T. G. M., Tetrahedron Lett. , 33, 1025 (1992). c) Schmidt D. D., Frommer W., Junge B., MullerL., Wingender W., Truscheit E., Naturwissenschaften, 64, 535 (1977). d) Itoh J., Omoyo S., Shomura T., Ogino H., Iwamatsu K.,
Inouye S., J. Antibiot. , 34, 1424 (1981). e) Yokose K., Ogawa K., Sano T., Watanabe K., Maruyama H., Suhara Y., J. Antibiot. , 36,1157 (1983).
15 a) Saunier B., Kilker R. D., Tkacz J. S., Quaroni A., Herscovics A., J. Biol. Chem., 257 , 14155 (1982). b) Pan Y. T., Hori H., SaulR., Sanford B. A., Molyneux R. J., Elbein A. D., Biochemistry , 22, 3975 (1983).
16 a) Atsumi S., Nosaka C., Ochi Y., Iinuma H., Umezawa K., Cancer Research , 53, 4896 (1993). b) Tatsuta K., Niwata Y., Umezawa.K., Toshima K., Nakata M., Carbohydr. Res., 222 , 189 (1991).
17 a) Tatsuta K., Niwata Y., Umezawa K., Toshima K., Nakata M., Tetrahedron Lett., 31, 1171 (1990). b) Tatsuta K., Niwata Y.,Umezawa K., Toshima K., Nakata M., J. Antibiot. , 44, 912 (1991). c) Vincent W. F. T., Fung P. H., Wong Y. S., Shing T. K. M.,Tetrahedron: Asymmetry, 5 , 1353 (1994). d) Nakata M., Chong M. C., Niwata Y., Toshima K., Tatsuta K., J. Antibiot. , 46, 1919(1993). e) Moritz V., Vogel P., Tetrahedron Lett. , 33, 5243 (1992). f) Shing T. K. M., Tai V. W. F., J. Chem. Soc., Perkin Trans. 1,2017 (1994). g) Schlessinger R. H., Bergstrom C. P., J. Org. Chem. , 60, 16 (1995). h) Mcdevitt R. E., Fraser-Reid B., J. Org.Chem., 59, 3250 (1994). i) Shing T. K. M., Tai V. W. F., J. Chem. Soc., Chem. Commun. , 995 (1993). j) Akiyama T., Ohnari M.,Shima H., Ozaki S., Synlett, 831 (1991).
18 Semeria D., Philippe M., Delaumeny J.-M., Sepulchre A.-M., Gero S. D., Synthesis, 710 (1983).
19 Gemal A. L., Luche J. L., J. Am. Chem. Soc., 103 , 5454 (1981).
20 Eliel E. L., Allinger N. L., Angyal S. J., Morrison G. A., Conformational Analysis., Wiley: New York. 1965, 352.
21 a) Pelter A., Bugden G., Rosser R., Tetrahedron Lett., 26, 5097 (1985). b) Pelter A., Singaram B., Warren L., Wilson J. W.,Tetrahedron, 49, 2965 (1993).
22 Similar analysis was conducted by Frost et al. Montchamp J. L., Migarud M. E., Frost J. W., J. Org. Chem., 58, 7679 (1993).
23 Data not shown.
24 a) Takahashi H., Kittaka H., Ikegami S., Tetrahedron Lett. , 39, 9703 (1998). b) Takahashi H., Kittaka H., Ikegami S., TetrahedronLett. , 39, 9707 (1998).
25 a) Sasaki K., Loewus F. A., Plant Physiol., 69, 220 (1982). b) Sasaki K., Taylor I. E. P., Plant Cell Physiol., 25, 989 (1984). c)Sasaki K., Taylor I. E. P., Plant Physiol., 81, 493 (1986).
26 a) Michell R. H., Biochim Biophys. Acta, 415 , 81 (1975). b) Berridge M. J., Irvine R. F., Nature (London), 312 , 315 (1984).
27 a) Ley S. V., Parra M., Redgrave A. J., Sternfeld F., Tetrahedron , 46, 4995 (1990). b) Bender S. L., Budhu R. J., J. Am. Chem.Soc., 11 3 , 9883 (1991) c) Ley S. V., Pure. Appl. Chem., 62, 2031 (1990). d) Bruzik K. S., Tsai M-D., J. Am. Chem. Soc., 11 4 , 6361(1992). e) Watanabe Y., Fujimoto T., Shinohara T., Ozaki S., J. Chem. Soc., Chem. Commun. , 428 (1991). f) Liu Y-C., Chen C-S.,Tetrahedron Lett. , 30, 1617 (1989). g) Ozaki S., Kondo Y., Nakahira H., Yamaoka S., Watanabe Y., Tetrahedron Lett. , 28, 4691(1987). h) Reddy K. M., Reddy K. K., Falck J. R., Tetrahedron Lett. , 38, 4951 (1997).
28 a) Steb H., Irvine R. F., Berridge M. J., Schulz I., Nature (London), 306 , 67 (1983). b) Majerus P. N., Conolly T. M., Deckmyn H.,Ross T. S., Bross T. E., Ishii H., Bansal V. S., Wilson D. B., Science, 234 , 1519 (1986). c) Putney J. W. Jr., Am. J. Physiol. , 252 ,G149 (1987).
29 a) Irvine R. F., Nature, 328 , 386 (1987). b) Irvine R. F., Moor R. M., Biochem. J., 240 , 917 (1986). c) Morris A. P., Gallacher D. V.,Irvine R. F., Petersen O. H., Nature, 330 , 653 (1987). d) Hill T. D., Dean N. M., Boynton A. L., Science , 242 , 1176 (1988). e)Changya L., Gallacher D. V., Irvine R. F., Petersen G. H., FEBS Lett. , 251 , 43 (1989). f) Joseph S. K., Hansen C. A., WilliamsonJ. R., Mol. Pharmacol., 36, 391 (1989). g) Ely J. A., Hunyady L., Baukal A. J., Catt K. J., Biochem. J., 268 , 333 (1990). h) GawlerP. J., Potter B. V. L., Nahorski S. R., Biochem. J., 272 , 519 (1990). i) Cullen P. J., Irvine R. F., Dawson A. P., Biochem. J., 271 , 549(1990). j) Molleman A., Hoiting B., Duin M., Akker J. van den, Nelemans A., Martog A. D., J. Biol. Chem., 266 , 5658 (1991).
30 Takenawa T. (ed), New development in signal transduction researches. Yodosha Co., Ltd. Tokyo (1993)
31 a) Kowarski A. R., Sarel S., J. Org. Chem., 38, 117 (1973). b) Mandel M., Hudlicky T., J. Chem. Soc., Perkin Trans. 1, 741 (1993).c) Balci M., Sütbeyaz Y., Secen H., Tetrahedron, 46, 3715 (1990). d) Billington D. C., Chem. Soc. Rev., 18, 83 (1989). e) AngyalS. J., Odier L., Tate M. E., Carbohydr. Res., 266 , 143 (1995). f) Angyal S. J., Hickman R. J., Carbohydr. Res., 20, 97 (1971).
32 Only myo -inositols have been synthesized by Ferrier (II) reaction using mercurate. Bender S. L., Budhu R. J., J. Am. Chem. Soc.,113 , 9883 (1991).
33 Yamauchi N., Terachi T., Eguchi T., Kakinuma K., Tetrahedron, 50, 4125 (1994).
34 Evans D. A., Chapman K. T., Carreira, E. M., J. Am. Chem. Soc. , 110 , 3560 (1988).
35 a) Sato K., Yoshitomo A., Takai Y., Bull. Chem. Soc. Jpn., 70, 885 (1997). b) Torisawa Y., Okabe H., Ikegami S., Chem. Lett., 1555(1984).
36 a) Angyal S. J., Ordier L., Carbohydr. Res., 100 , 43 (1982). b) Sasaki K., Hicks K. B., Nagahashi G., Carbohydr. Res., 183 , 1(1988).
37 Inositol (3,4,5) triphosphate. Lindon J. C., Baker D. J., Farrant R. D., Williams J. M., Biochem. J., 233 , 275 (1986).
38 Inositol (1,3,4,5) tetraphosphate. a) Lindon J. C., Baker D. J., Williams J. M., Irvine R. F., Biochem. J. , 244 , 591 (1987). b) CerdanS., Hansen C. A., Johanson R., Inubushi T., Williams J. R., J. Biol. Chem. , 261 , 14676 (1986).
39 a) Takahashi H., Hitomi Y., Iwai Y., Ikegami S., J. Am. Chem. Soc., 122 , 2995 (2000). b) Takahashi, H.; Iwai, Y.; Hitomi, Y.; Ikegami,S., Org. Lett., 4 , 2401, (2002).
40 a) Umezawa H., Maeda K., Takeuchi T., Okami Y., J. Antibiot. Ser. A , 19, 200 (1966). b) Sina? P., Jacquinet J.-C., CarbohydrateResearch, 132 , C5 (1984).
41 a) Mattingly P. G., Kerwin J. F. Jr., Miller M. J., J. Am. Chem. Soc., 101 , 3983 (1979). b) Morrison M. A., Miller M. J., J. Org. Chem.,48, 4421 (1983). c) Miller M. J., Mattingly P. G., Tetrahedron , 39, 2563 (1983). d) Farouz F., Miller M. J., Tetrahedron Lett. , 32,3305 (1991).
42 a) Miller M. J., Mattingly P. G., Morrison M. A., Kerwin J. F. Jr., J. Am. Chem. Soc., 102 , 7026 (1980). b) Bose A. K., Sahu D. P.,Manhas M. S., J. Org. Chem. , 46, 1229 (1981). c) Krook M. A., Miller M. J., J. Org. Chem. , 50, 1126 (1985). d) Galéotti N.,Montagne C., Poncet J., Jouin P., Tetrahedron Lett. , 33, 2807 (1992). e) Koppel I., Koppel J., Koppel I., Leito I., Pihl V., Wallin A.,Grehn L., Ragnarsson U., J. Chem. Soc., Perkin Trans. 2 , 655 (1993).
43 a) Houghton R. P., Williams C. S., Tetrahedron Lett. , 8 , 3929 (1967). b) Levin J. I., Turos E., Weinreb S. M., Synth. Commun., 12,989 (1982). c) Lesimple P., Bigg D. C. H., Synthesis, 306 (1991).
44 For L-RNA (a) Ashley, G. W., J. Am. Chem. Soc. , 114 , 9731 (1992). For L-DNA (b) Damha, M. J.; Giannaris, P. A.; Marfey, P.,Biochemistry , 33, 7877 (1994). (c) Hashimoto, Y.; Iwanami, N.; Fujimori, S.; Shudo, K., J. Am. Chem. Soc., 115 , 9883 (1993). (d)Fujimori, S.; Shudo, K.; Hashimoto, Y., J. Am. Chem. Soc. , 112 , 7436 (1990).
45 a) W. D. Ollis, C. Smith, D. E. Wright, Tetrahedron, 35, 105 (1979); b) D. E. Wright, Tetrahedron, 35, 1207 (1979).
46 a) H. Ohtake, T. Iimori, S. Ikegami, Tetrahedron Lett. , 38, 3413 (1997); b) H. Ohtake, T. Iimori, M. Shiro, S. Ikegami, Heterocycles ,
47, 685 (1998); c) H. Ohtake, N. Ichiba, M. Shiro, S. Ikegami, J. Org. Chem., 65, 8164 (2000).47 M. Kurihara, N. Miyata, Chem. Lett., 1995, 263.
48 a) H. Ohtake, T. Iimori, S. Ikegami, Tetrahedron Lett. , 38, 3413 (1997); b) H. Ohtake, T. Iimori, M. Shiro, S. Ikegami, Heterocycles ,47, 685 (1998); c) H. Ohtake, N. Ichiba, M. Shiro, S. Ikegami, J. Org. Chem., 65, 8164 (2000).
49 F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendricson, W. C. Still, J. Comput.Chem. , 11, 440 (1990).
50 a) I. Kolossváry, W. C. Guida, J. Am. Chem. Soc., 118 , 5011 (1996); b) G. Cheng, W. C. Guida, W. C. Still, J. Am. Chem. Soc., 111 ,4379 (1989).
51 T. Yoshimura, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 42, 514 (1984).
52 a) R. U. Lemieux, “Molecular Rearrangements”, ed. by P. De Mayo, Interscience, New York, 1964; b) A. J. Kirby, “The AnomericEffect and Related Stereoelectronic Effects at Oxygen”, Springer Verlag, Berlin, 1983; c) I. Tvaroska, T. Bleha, “Advances inCarbohydrate Chemistry and Biochemistry”, Vol. 47, eds. by R. S. Tipson, D. Horton, Academic Press, San Diego, 1989, p. 45.
53 a) K. Pihlaja, J. Heokkli*, Acta Chem. Scand. , 21, 2390 (1967); b) E. L. Eliel, F. W. Nader, J. Am. Chem. Soc., 92, 584 (1970); c)U. Salzner, P. v. R. Schleyer, J. Org. Chem., 59, 2138 (1994).
54 a) T. Iimori, H. Ohtake, S. Ikegami, Tetrahedron Lett. , 38, 3415 (1997); b) H. Ohtake, T. Iimori, S. Ikegami, Synlett, 1998, 1420; c)H. Ohtake, N. Ichiba, S. Ikegami, J. Org. Chem., 65, 8171 (2000).
55 a) K. Toshima, K. Tatsuta, Chem. Rev., 93, 1503 (1993); b) G. -J. Boons, Tetrahedron, 52, 1095 (1996); c) S. J. Danishefsky, M.T. Bilodeau, Angew. Chem., Int. Ed. Engl., 35, 1380 (1996); d) S. Hashimoto, Y. Honda, Y. Yanagitani, M. Nakajima, S. Ikegami,Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 53, 620 (1995).
56 a) S. S. Bhattacharjee, P. A. J. Gorin, <st1:country-region w:st="on">Can. J. Chem., 47, 1195 (1969); b) A. Lipták, I. Jodál, P. Nánási, Carbohydr. Res., 44, 1(1975).
57 a) P. J. Garegg, H. Hultberg, S. Wallin, Carbohydr. Res., 108 , 97 (1982); b) R. Johansson, B. Samuelsson, J. Chem. Soc., PerkinTrans. 1 , 1984, 2371.
58 P. Deslongchamps, R. Chénevert, R. J. Taillefer, C. Moreau, J. K. Saunders, Can. J. Chem., 53, 1601 (1975).
59 Other step-wise b-mannosylation methods: a) A. Dan, A. Y. Ito, T. Ogawa, Tetrahedron Lett. , 36, 7487 (1995). idem., J. Org.Chem., 60, 4680 (1995); b) F. Barresi, O. Hindsgaul, <st1:country-region w:st="on">Can. J. Chem., 72, 1447 (1994); c) G. Stork, J. J. La Clair, J. Am. Chem.Soc., 118 , 247 (1996); d) F. W. Lichtenthaler, T. Schneider-Adams, S. Immel, J. Org. Chem., 59, 6735 (1994).
60 N. Hada, I. Ohtsuka, M. Sugita, T. Takeda, Tetrahedron Lett. , 41, 9065 (2000).
61 a) H. Ohtake, S. Ikegami, Org. Lett., 2, 457 (2000); b) H. Ohtake, X. L. Li, M. Shiro, S. Ikegami, Tetrahedron, 56, 7109 (2000).
62 Y. Naruse, H. Yamamoto, Tetrahedron Lett. , 27, 1363 (1986).
63 Mukaiyama, T.; Banno, K.; Narasaka, K., J. Am. Chem. Soc., 96, 7503 (1974).
64 Kobayashi, S.; Nagayama, S.; Busujima, T., J. Am. Chem. Soc., 120 , 8287 (1998).
65 H. Fukase, S. Horii, J. Org. Chem., 57, 3642 (1992).
66 a) Y. Kameda, M. Asano, M. Yoshikawa, M. Takeuchi, T. Yamaguchi, K. Matsui, S. Horii, H. Fukase, J. Antibiot. , 37, 1301 (1984);b) S. Horii, H. Fukase, Y. Kameda, Carbohydr. Res., 140 , 185 (1985).
67 a) H. Paulsen, F. R. Heiker, Liebigs Ann. Chem. , 1981, 2180; b) N. Sakairi, H. Kuzuhara, Tetrahedron Lett. , 23, 5327 (1982); c) S.Ogawa, N. Chida, T. Suami, J. Org. Chem., 48, 1203 (1983); d) R. R. Schmidt, A. K?hn, Angew. Chem., Int. Ed. Engl., 26, 482(1987); e) M. Yoshikawa, B. C. Cha, T. Nakae, I. Kitagawa, Chem. Pharm. Bull., 36, 3714 (1988); f) F. Nicotra, L. Panza, F.Ronchetti, G. Russo, Gazz. Chim. Ital., 11 9 , 577 (1989); g) T. K. Park, S. J. Danishefsky, Tetrahedron Lett. , 35, 2667 (1994); h) T.K. M. Shing, L. H. Wan, L. H., J. Org. Chem., 61, 8468 (1996); i) B. M. Trost, L. S. Chupak, T. Lübbers, J. Am. Chem. Soc., 120 ,1732 (1998); j) S. Ogawa, C. Uchida, T. Ohhira, Carbohydr. Lett., 3 , 277, (1999).
68 O.22) Fukase, Yuki Gosei Kagaku Kyokaishi (J. Synth. Org. Chem. Jpn.), 55, 920 (1997); b) H. Fukase, S. Horii, J. Org. Chem.,57, 3642 (1992); c) H. Fukase, S. Horii, J. Org. Chem., 57, 3651 (1992); d) S. Horii, H. Fukase, T. Matsuo, Y. Kameda, N. Asano,K. Matsui, J. Med. Chem. , 29, 1038 (1986).
注:本文為提供者整理翻譯的,由于知識所限,錯誤在所難免,敬請原諒。如有問題可以查找原文。
快速導航
化學品: a | b | c | d | e | f | g | h | i | j | k | l | m | n | o | p | q | r | s | t | u | v | w | x | y | z | 0 | 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9
關于物競
物競數據庫是一個全面、專業、專注,并且免費的中文化學品信息庫,為學生、學者、化學品研究機構、檢測機構、化學品工作者提供專業的化學品平臺進行交流。
數據庫采用全中文化服務,完全突破了中英文在化學物質命名、化學品俗名、學名等方面的差異,所提供的數據全部中文化,更方便國內從事化學、化工、材料、生物、環境等化學相關行業的工作人員查詢使用。
關注我們

-
微信賬號:物競化學品數據庫

-
微博賬號:wjhxp
聯系我們
上海市延長路149號上海大學科技園412室
公司總機: 021-56389801
訂購電話: 4007001514
傳真電話: 021-56389802
客服電話: 021-56332350
電子郵件: wingch@basechem.org
 滬公網安備 31010602001115號
滬公網安備 31010602001115號